Publicado originalmente el 3 de mayo de 2017 y actualizado por última vez el 24 de septiembre de 2020
Este post ha sido elaborado por el bloguero invitado, miembro del Consejo Asesor de Addgene y científico del Instituto Broad, John Doench.
La tecnología CRISPR ha facilitado más que nunca tanto la ingeniería de ediciones específicas de ADN como la realización de pantallas funcionales para identificar los genes implicados en un fenotipo de interés. En esta entrada del blog se analizarán las diferencias entre estos enfoques y se proporcionará información actualizada sobre la mejor manera de diseñar gRNAs. También puede encontrar ARNg validados para su próximo experimento en la base de datos de secuencias de ARNg validadas de Addgene. Una discusión más extensa de estos temas se puede encontrar en dos artículos de revisión recientes (Doench et al., 2017, y Hanna et al., 2020) y las referencias en los mismos.

Nom nom nom. Me encantan los GTACAGCCTG. Cómic de Maya Kostman.
Consideraciones importantes antes de empezar un experimento con CRISPR
El martillo, la sierra de calar y la llave inglesa son herramientas estupendas, pero la que se utilice, por supuesto, depende de lo que se intente hacer: no hay una herramienta «mejor» entre ellas. Aunque esto parece obvio, es importante recordar que lo mismo es cierto cuando se diseñan gRNAs para el uso de la tecnología CRISPR – el «mejor» gRNA depende en gran medida de lo que se está tratando de hacer: knockout de genes, una edición de base específica, o la modulación de la expresión génica.
 La ubicación y la secuencia son consideraciones importantes para el diseño de sus gRNAs. En el caso de los indels, no es tan importante la ubicación en el gen al que se dirigen, pero sí es importante que la secuencia de su ARNg esté diseñada para ser altamente activa y reducir los objetivos. Para CRISPRa y CRISPRi, estas consideraciones tienen aproximadamente la misma importancia (el objetivo debe estar cerca del TSS, pero puede preocuparse menos por la optimización de la secuencia porque generalmente tiene menos secuencias para elegir). Por último, para HDR, la ubicación es mucho más importante porque usted tiene que apuntar dentro de ~ 30 nt de su propuesta de edición, lo que significa que hay tan pocos gRNAs para elegir que las preferencias de secuencia deben ser ignoradas en gran medida.
La ubicación y la secuencia son consideraciones importantes para el diseño de sus gRNAs. En el caso de los indels, no es tan importante la ubicación en el gen al que se dirigen, pero sí es importante que la secuencia de su ARNg esté diseñada para ser altamente activa y reducir los objetivos. Para CRISPRa y CRISPRi, estas consideraciones tienen aproximadamente la misma importancia (el objetivo debe estar cerca del TSS, pero puede preocuparse menos por la optimización de la secuencia porque generalmente tiene menos secuencias para elegir). Por último, para HDR, la ubicación es mucho más importante porque usted tiene que apuntar dentro de ~ 30 nt de su propuesta de edición, lo que significa que hay tan pocos gRNAs para elegir que las preferencias de secuencia deben ser ignoradas en gran medida.
El martillo: Eliminación de genes por NHEJ
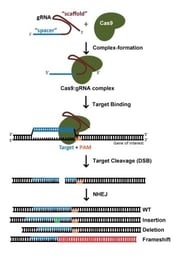 La eliminación de genes con la tecnología CRISPR se consigue normalmente mediante roturas de dsDNA mediadas por Cas9: después de un corte, la naturaleza propensa a errores de la unión de extremos no homólogos (NHEJ) a menudo conduce a la generación de indels y, por tanto, a cambios de marco que interrumpen la capacidad de codificación de proteínas de un locus. Cuando se utiliza S. pyogenes Cas9 (SpCas9), los sitios objetivo potenciales son ambos y , ya que es igualmente eficaz dirigirse a la cadena codificante o no codificante del ADN. Como regla general, evitamos los sitios objetivo que codifican aminoácidos cerca de la terminación N’ de la proteína, con el fin de mitigar la capacidad de la célula para utilizar un ATG alternativo aguas abajo del codón de inicio anotado. Del mismo modo, evitamos los sitios objetivo que codifican aminoácidos cerca del extremo C’ de la proteína, para maximizar las posibilidades de crear un alelo no funcional. Para un gen de 1 kilobase, dado que los sitios objetivo potenciales se producen ~1 de cada 8 nucleótidos, restringir los ARNg a un 5 – 65% de la región de codificación de la proteína aún dará como resultado muchas docenas de ARNg para elegir. Con tantas posibilidades, la elección de un ARNg con una secuencia optimizada es de importancia primordial (más sobre esto a continuación).
La eliminación de genes con la tecnología CRISPR se consigue normalmente mediante roturas de dsDNA mediadas por Cas9: después de un corte, la naturaleza propensa a errores de la unión de extremos no homólogos (NHEJ) a menudo conduce a la generación de indels y, por tanto, a cambios de marco que interrumpen la capacidad de codificación de proteínas de un locus. Cuando se utiliza S. pyogenes Cas9 (SpCas9), los sitios objetivo potenciales son ambos y , ya que es igualmente eficaz dirigirse a la cadena codificante o no codificante del ADN. Como regla general, evitamos los sitios objetivo que codifican aminoácidos cerca de la terminación N’ de la proteína, con el fin de mitigar la capacidad de la célula para utilizar un ATG alternativo aguas abajo del codón de inicio anotado. Del mismo modo, evitamos los sitios objetivo que codifican aminoácidos cerca del extremo C’ de la proteína, para maximizar las posibilidades de crear un alelo no funcional. Para un gen de 1 kilobase, dado que los sitios objetivo potenciales se producen ~1 de cada 8 nucleótidos, restringir los ARNg a un 5 – 65% de la región de codificación de la proteína aún dará como resultado muchas docenas de ARNg para elegir. Con tantas posibilidades, la elección de un ARNg con una secuencia optimizada es de importancia primordial (más sobre esto a continuación).
El rompecabezas: Edición por HDR, edición de bases y edición de primos
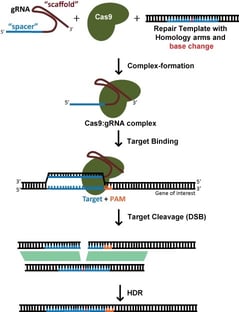
Para una edición específica, como la inserción de una etiqueta fluorescente o la introducción de una mutación específica, uno generalmente se basa en la reparación dirigida por homología (HDR) para incorporar nueva información en el ADN. Esto también requiere una plantilla de ADN exógena. Sin embargo, la HDR es un proceso de muy baja eficiencia y suele implicar la necesidad de clonar una sola célula y el posterior cribado de las ediciones exitosas. Se trata de un proceso que requiere mucho tiempo y que no debe llevarse a cabo a la ligera. De hecho, lograr realmente el estándar de oro requiere no una, sino dos rondas de clonación de células individuales – como control, uno debe revertir la edición de nuevo al original con el fin de demostrar que el fenotipo se debió realmente a la edición prevista en lugar de alguna variante de pasajeros que vino junto con el clon de células individuales (aunque esto se hace raramente).
Cuando se apunta a una ruptura de dsDNA para la HDR, la elección del sitio objetivo está mucho más limitada por la ubicación deseada de la edición; la eficiencia disminuye dramáticamente cuando el sitio de corte es >30nt de los extremos proximales de la plantilla de reparación (Yang et al., 2013). Esto significa que, para la edición de genes, suele haber pocos ARNg potenciales. Mientras que SpCas9, con una preferencia de PAM de NGG, sigue siendo la enzima Cas más utilizada, el desarrollo de las enzimas SaCas9, NmeCas9, Cas12a y sus variantes de ingeniería ofrece opciones adicionales de PAM que pueden ampliar en gran medida las opciones de gRNA.
Dos tecnologías más nuevas ofrecen una alternativa a la HDR para introducir ediciones. Las mismas restricciones de localización son aún más exquisitas para el llamado editor de bases Cas9, que realiza cambios en el ADN en ausencia de roturas de dsDNA (Rees et al., 2018). Para los editores de bases C>T y A>G, la edición prevista debe estar en una ventana de 5 a 10 nt en relación con la PAM, y las ediciones secundarias son posibles si hay otra C o A objetivo en la ventana. Otra tecnología, la edición primaria (revisada en Anzalone et al., 2020), no se limita a las transiciones de un solo nucleótido, pero sigue requiriendo una PAM cercana, aunque todavía son los primeros días para esta tecnología, y el usuario puede necesitar optimizar numerosos parámetros para generar la edición deseada.
La llave inglesa: Activación e inhibición de genes mediante CRISPRa y CRISPRi
Por último, para modular la expresión génica a nivel de la transcripción -tecnologías CRISPRa (activación) y CRISPRi (inhibición)- se dirige un Cas9 muerto por nucleasa (dCas9) cerca del promotor de un gen diana. En este caso, la ventana objetivo no es tan amplia como en el caso del knockout por corte CRISPR. En el caso de CRISPRa, lo más eficaz es dirigirse a una ventana de ~100nt aguas arriba del sitio de inicio de la transcripción (TSS), mientras que en el caso de CRISPRi, una ventana de ~100nt aguas abajo del TSS es la que proporciona la mayor actividad. Por lo tanto, un gen determinado sólo tendrá una docena de ARNg para elegir en la ubicación óptima. También es importante tener una buena información sobre la ubicación exacta del TSS. Diferentes bases de datos anotan el TSS de diferentes maneras, y se ha demostrado que la base de datos FANTOM, que se basa en CAGE-seq para capturar directamente la tapa del ARNm, proporciona el mapeo más preciso (Radzisheuskaya et al., 2016). En este caso, la ubicación y la secuencia tienen aproximadamente la misma importancia en el diseño – una secuencia optimizada hará poco si está en el lugar equivocado, pero debido a que la ventana de destino es más estrecha, hay menos ARNg para elegir, y por lo tanto una secuencia óptima puede no estar disponible.
Prediciendo la eficacia del gRNA
Nosotros y otros hemos examinado la capacidad de utilizar características basadas en la secuencia y otras para nominar gRNAs que probablemente sean activos, no sólo para SpCas9 sino también para algunas otras enzimas Cas. Parece ser que no existe un sistema de puntuación universal para seleccionar un ARNg, ya que el método de producción de la guía (sintético, transcripción in vitro o entrega lentiviral) puede afectar a la precisión de una puntuación predictiva, así como a los aspectos dinámicos de la diana (por ejemplo, la accesibilidad debida al estado de la cromatina). Ninguna predicción computacional es perfecta, pero esto puede disminuir el número de guías que uno necesita probar en el laboratorio.
Es importante que, para cualquier modificación de interés, no sería prudente sacar conclusiones sobre la base de la actividad de un solo ARNg, y por lo tanto la diversidad de ARNg a través de un gen debe ser examinada siempre que sea posible cuando se utilizan enfoques de knockout o de modulación transcripcional.
Evitar los efectos fuera del objetivo

Es importante tener en cuenta la actividad fuera del objetivo de los ARNg. Aunque se ha establecido el panorama básico de desajustes que, sin embargo, pueden dar lugar a actividad, y puede utilizarse para identificar los sitios que probablemente den lugar a un efecto fuera de objetivo, no hay suficientes datos para predecir completamente qué sitios mostrarán o no niveles apreciables de modificación. La secuenciación del genoma completo de las células modificadas por CRISPR indica que las consecuencias de la actividad fuera del objetivo, al menos para las condiciones experimentales utilizadas, no dieron lugar a mutaciones detectables (Veres et al., 2014). Cuando se trabaja con clones unicelulares, los autores señalan que «la heterogeneidad clonal puede representar un obstáculo más serio para la generación de líneas celulares verdaderamente isogénicas que los efectos off-target mediados por las nucleasas.» Además, los conjuntos de datos a gran escala de cientos de pantallas genéticas que utilizan bibliotecas de todo el genoma han mostrado una alta concordancia entre las diferentes secuencias que se dirigen al mismo gen, lo que sugiere que los efectos fuera del objetivo no abruman la verdadera señal en estos ensayos (Dempster et al., 2019). Una vez más, la estrategia experimental es clara: para cualquier gen de interés, se debe exigir que múltiples gRNAs de diferentes secuencias den lugar al mismo fenotipo para concluir que el fenotipo se debe a un efecto on-target.
Conclusiones
La selección de gRNAs para un experimento necesita equilibrar la maximización de la actividad on-target y la minimización de la actividad off-target, lo que suena obvio pero a menudo puede requerir decisiones difíciles. Por ejemplo, ¿sería mejor utilizar un ARNg menos activo que se dirija a un sitio realmente único en el genoma, o un ARNg más activo con un sitio objetivo adicional en una región del genoma sin función conocida? Para la creación de modelos celulares estables que se van a utilizar para un estudio a largo plazo, la primera puede ser la mejor opción. Sin embargo, para una biblioteca de todo el genoma para llevar a cabo pantallas genéticas, una biblioteca compuesta por el segundo probablemente sería más eficaz, siempre y cuando se tenga cuidado en la interpretación de los resultados al requerir múltiples secuencias dirigidas a un gen para puntuar con el fin de llamar a ese gen un éxito.
Esta es una época emocionante para la genómica funcional, con una lista cada vez más amplia de herramientas para sondear la función de los genes. Las mejores herramientas son tan buenas como la persona que las utiliza, y el uso adecuado de la tecnología CRISPR siempre dependerá del diseño experimental cuidadoso, la ejecución y el análisis.

¡Muchas gracias a nuestro bloguero invitado John Doench!
 John Doench es el Director de R&D en la Plataforma de Perturbación Genética en el Instituto Broad y ha trabajado con muchos Addgenies para ayudar a mejorar la comprensión, curación y explicación de nuestros recursos CRISPR. Le gustan mucho los ARN pequeños.
John Doench es el Director de R&D en la Plataforma de Perturbación Genética en el Instituto Broad y ha trabajado con muchos Addgenies para ayudar a mejorar la comprensión, curación y explicación de nuestros recursos CRISPR. Le gustan mucho los ARN pequeños.
Anzalone AV, Koblan LW, Liu DR (2020) Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38:824-844 . https://doi.org/10.1038/s41587-020-0561-9
Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H, Allen F, Gonçalves E, Shepherd R, Doench JG, Yusa K, Vázquez F, Parts L, Boehm JS, Golub TR, Hahn WC, Root DE, Garnett MJ, Tsherniak A, Iorio F (2019) Acuerdo entre dos grandes conjuntos de datos de dependencia de genes CRISPR-Cas9 pan-cáncer. Nat Commun 10: . https://doi.org/10.1038/s41467-019-13805-y
Doench JG (2017) ¿Estoy preparado para CRISPR? Una guía de usuario para pantallas genéticas. Nat Rev Genet 19:67-80 . https://doi.org/10.1038/nrg.2017.97
Hanna RE, Doench JG (2020) Diseño y análisis de experimentos CRISPR-Cas. Nat Biotechnol 38:813-823 . https://doi.org/10.1038/s41587-020-0490-7
Radzisheuskaya A, Shlyueva D, Müller I, Helin K (2016) La optimización de la posición del sgRNA mejora notablemente la eficiencia de la represión transcripcional mediada por CRISPR/dCas9. Nucleic Acids Res 44:e141-e141 . https://doi.org/10.1093/nar/gkw583
Rees HA, Liu DR (2018) Edición de bases: química de precisión en el genoma y el transcriptoma de las células vivas. Nat Rev Genet 19:770-788 . https://doi.org/10.1038/s41576-018-0059-1
Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K (2014) Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell 15:27-30 . https://doi.org/10.1016/j.stem.2014.04.020
Yang L, Guell M, Byrne S, Yang JL, De Los Ángeles A, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Ríos X, Huang P-Y, Daley G, Church G (2013) Optimización de la edición del genoma de células madre humanas sin cicatrices. Nucleic Acids Research 41:9049-9061 . https://doi.org/10.1093/nar/gkt555
Recursos en el blog de Addgene
- Escucha nuestro Podcast con John Doench
- Aprende cómo llevar a cabo cribados de bibliotecas agrupadas de CRISPR en todo el genoma
- Lee otras publicaciones del blog de CRISPR
.
0 comentarios