Originariamente pubblicato il 3 maggio 2017 e aggiornato il 24 settembre 2020
Questo post è stato contribuito da John Doench, blogger ospite, membro dell’Addgene Advisory Board e scienziato dell’Istituto Broad Institute.
La tecnologiaCRISPR ha reso più facile che mai sia l’ingegnerizzazione di modifiche specifiche del DNA che l’esecuzione di schermi funzionali per identificare i geni coinvolti in un fenotipo di interesse. Questo post del blog discuterà le differenze tra questi approcci e fornirà aggiornamenti su come progettare al meglio i gRNA. Puoi anche trovare gRNA convalidati per il tuo prossimo esperimento in Addgene’s Validated gRNA Sequence Datatable. Una discussione più estesa di questi argomenti può essere trovata in due recenti articoli di revisione (Doench et al., 2017, e Hanna et al., 2020) e relativi riferimenti.

Nom nom nom. Io amo i gRNA! Comic di Maya Kostman.
Considerazioni importanti prima di iniziare un esperimento con CRISPR
Il martello, il seghetto e la chiave inglese sono tutti ottimi strumenti, ma quale usi, ovviamente, dipende da cosa stai cercando di fare – non c’è uno strumento “migliore” tra loro. Anche se questo sembra ovvio, è importante ricordare che lo stesso è vero quando si progettano i gRNA per l’utilizzo della tecnologia CRISPR – il “migliore” gRNA dipende molto da quello che si sta cercando di fare: knockout genico, una modifica specifica della base, o la modulazione dell’espressione genica.
 La posizione e la sequenza sono considerazioni importanti per la progettazione dei vostri gRNA. Per gli indel, non è così importante quale posizione nel gene prendi di mira, ma è importante che la tua sequenza di gRNA sia progettata per essere altamente attiva e ridurre gli off target. Per CRISPRa e CRISPRi, queste considerazioni sono all’incirca della stessa importanza (il target dovrebbe essere vicino al TSS, ma ci si può preoccupare meno dell’ottimizzazione della sequenza perché generalmente si hanno meno sequenze tra cui scegliere). Infine, per HDR, la posizione è molto più importante perché devi mirare entro ~ 30 nt della tua proposta di modifica, il che significa che ci sono così pochi gRNA da scegliere che le preferenze di sequenza devono essere ampiamente ignorate.
La posizione e la sequenza sono considerazioni importanti per la progettazione dei vostri gRNA. Per gli indel, non è così importante quale posizione nel gene prendi di mira, ma è importante che la tua sequenza di gRNA sia progettata per essere altamente attiva e ridurre gli off target. Per CRISPRa e CRISPRi, queste considerazioni sono all’incirca della stessa importanza (il target dovrebbe essere vicino al TSS, ma ci si può preoccupare meno dell’ottimizzazione della sequenza perché generalmente si hanno meno sequenze tra cui scegliere). Infine, per HDR, la posizione è molto più importante perché devi mirare entro ~ 30 nt della tua proposta di modifica, il che significa che ci sono così pochi gRNA da scegliere che le preferenze di sequenza devono essere ampiamente ignorate.
Il martello: Gene knockout by NHEJ
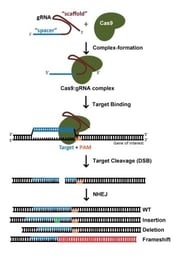 Gene knockout con la tecnologia CRISPR è solitamente realizzato da rotture di dsDNA mediate da Cas9: a seguito di un taglio, la natura soggetta a errori della non-homologous end joining (NHEJ) spesso porta alla generazione di indelebili e quindi frameshifts che interrompono la capacità di codifica delle proteine di un locus. Quando si usa S. pyogenes Cas9 (SpCas9), i potenziali siti di destinazione sono sia e , in quanto è altrettanto efficace per colpire il filamento codificante o non codificante del DNA. Come regola generale, evitiamo i siti di destinazione che codificano per gli aminoacidi vicino al termine N’ della proteina, al fine di mitigare la capacità della cellula di utilizzare un ATG alternativo a valle del codone di inizio annotato. Allo stesso modo, evitiamo i siti di destinazione che codificano per gli aminoacidi vicino alla terminazione C’ della proteina, per massimizzare le possibilità di creare un allele non funzionale. Per un gene di 1 kilobase, dal momento che i potenziali siti di destinazione si verificano ~ 1 ogni 8 nucleotidi, limitando i gRNA al 5 – 65% della regione codificante della proteina risulterà ancora in molte decine di gRNA da scegliere. Con così tante possibilità, la scelta di un gRNA con una sequenza ottimizzata è di primaria importanza (più avanti su questo).
Gene knockout con la tecnologia CRISPR è solitamente realizzato da rotture di dsDNA mediate da Cas9: a seguito di un taglio, la natura soggetta a errori della non-homologous end joining (NHEJ) spesso porta alla generazione di indelebili e quindi frameshifts che interrompono la capacità di codifica delle proteine di un locus. Quando si usa S. pyogenes Cas9 (SpCas9), i potenziali siti di destinazione sono sia e , in quanto è altrettanto efficace per colpire il filamento codificante o non codificante del DNA. Come regola generale, evitiamo i siti di destinazione che codificano per gli aminoacidi vicino al termine N’ della proteina, al fine di mitigare la capacità della cellula di utilizzare un ATG alternativo a valle del codone di inizio annotato. Allo stesso modo, evitiamo i siti di destinazione che codificano per gli aminoacidi vicino alla terminazione C’ della proteina, per massimizzare le possibilità di creare un allele non funzionale. Per un gene di 1 kilobase, dal momento che i potenziali siti di destinazione si verificano ~ 1 ogni 8 nucleotidi, limitando i gRNA al 5 – 65% della regione codificante della proteina risulterà ancora in molte decine di gRNA da scegliere. Con così tante possibilità, la scelta di un gRNA con una sequenza ottimizzata è di primaria importanza (più avanti su questo).
Il puzzle: Editing tramite HDR, editing delle basi e prime editing
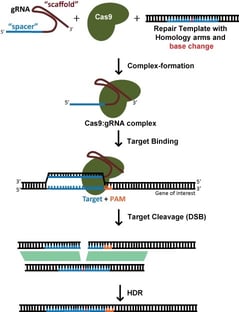
Per una modifica specifica, come l’inserimento di un tag fluorescente o l’introduzione di una mutazione specifica, ci si affida generalmente all’homology directed repair (HDR) per incorporare nuove informazioni nel DNA. Anche questo richiede un modello di DNA esogeno. L’HDR, tuttavia, è un processo a bassissima efficienza, e di solito comporta la necessità di clonare una singola cellula e di effettuare uno screening successivo per le modifiche riuscite. Questo è un processo che richiede molto tempo e non dovrebbe essere intrapreso alla leggera! Infatti, raggiungere veramente il gold standard richiede non uno ma due cicli di clonazione di cellule singole – come controllo, si dovrebbe riportare la modifica all’originale per dimostrare che il fenotipo era davvero dovuto alla modifica prevista piuttosto che a qualche variante passeggera che è arrivata con il clone di una singola cellula (anche se questo viene fatto raramente).
Quando si punta a una rottura del dsDNA per l’HDR, la scelta del sito di destinazione è molto più vincolata dalla posizione desiderata di modifica; l’efficienza diminuisce drasticamente quando il sito di taglio è >30nt dalle estremità prossimali del template di riparazione (Yang et al, 2013). Questo significa che, per l’editing genico, di solito ci sono pochi potenziali gRNA. Mentre SpCas9, con una preferenza PAM di NGG, è ancora l’enzima Cas più utilizzato, lo sviluppo di SaCas9, NmeCas9, gli enzimi Cas12a, e le loro varianti ingegnerizzate offre ulteriori opzioni PAM che possono espandere notevolmente le opzioni gRNA.
Due nuove tecnologie offrono un’alternativa a HDR per introdurre modifiche. Gli stessi vincoli locali sono ancora più squisiti per il cosiddetto base editor Cas9, che apporta modifiche al DNA in assenza di rotture del dsDNA (Rees et al., 2018). Per gli editor di basi C>T e A>G, la modifica prevista deve essere in una finestra di 5 – 10 nt relativa al PAM, e sono possibili modifiche accessorie se c’è un altro target C o A nella finestra. Un’altra tecnologia, il prime editing (recensito in Anzalone et al., 2020). non è limitato a singole transizioni nucleotidiche ma richiede ancora un PAM vicino, anche se questi sono ancora i primi giorni per questa tecnologia, e l’utente può avere bisogno di ottimizzare numerosi parametri per generare la modifica desiderata.
La chiave: Attivazione e inibizione dei geni con CRISPRa e CRISPRi
Infine, per modulare l’espressione genica a livello della trascrizione – le tecnologie CRISPRa (attivazione) e CRISPRi (inibizione) – una Cas9 muta nucleasi (dCas9) è diretta vicino al promotore di un gene bersaglio. Qui, la finestra di destinazione non è così ampia come per il knockout tramite taglio CRISPR. Per CRISPRa, è più efficace puntare una finestra di ~100nt a monte del sito di inizio della trascrizione (TSS), mentre per CRISPRi, una finestra di ~100nt a valle del TSS dà la maggior attività. Così, un dato gene avrà solo una dozzina di gRNA tra cui scegliere nella posizione ottimale. È anche importante avere buone informazioni sulla posizione esatta del TSS. Diversi database annotano il TSS in modi diversi, ed è stato dimostrato che il database FANTOM, che si basa sul CAGE-seq per catturare direttamente il cappuccio dell’mRNA, fornisce la mappatura più accurata (Radzisheuskaya et al., 2016). In questo caso, la posizione e la sequenza sono di circa la stessa importanza nella progettazione – una sequenza ottimizzata farà poco se è nel posto sbagliato, ma perché la finestra di destinazione è più stretta, ci sono meno gRNA da scegliere, e quindi una sequenza ottimale potrebbe non essere disponibile.
Predicendo l’efficacia dei gRNA
Noi e altri abbiamo esaminato la capacità di usare la sequenza e altre caratteristiche per nominare i gRNA che probabilmente saranno attivi, non solo per SpCas9 ma anche per alcuni altri enzimi Cas. Sembra che non esista un sistema di punteggio universale per la selezione di un gRNA, poiché il metodo di produzione della guida (sintetico, trascrizione in vitro o consegna lentivirale) può influenzare la precisione di un punteggio predittivo, così come gli aspetti dinamici del bersaglio (ad esempio l’accessibilità dovuta allo stato della cromatina). Nessuna previsione computazionale è mai perfetta, ma questo può diminuire il numero di guide da testare in laboratorio.
Importante, per qualsiasi modifica di interesse, non sarebbe saggio trarre conclusioni sulla base dell’attività di un singolo gRNA, e quindi la diversità di gRNA in un gene dovrebbe essere esaminata quando possibile quando si usano approcci di knockout o modulazione trascrizionale.
Evitare gli effetti off-target

L’attività off-target dei gRNA è importante da considerare. Mentre il panorama di base dei mismatch che possono comunque portare all’attività è stato stabilito, e può essere usato per identificare i siti che probabilmente daranno luogo a un effetto off-target, non ci sono abbastanza dati per prevedere completamente quali siti mostreranno o meno livelli apprezzabili di modifica. Il sequenziamento dell’intero genoma di cellule modificate da CRISPR indica che le conseguenze dell’attività off-target, almeno per le condizioni sperimentali utilizzate, non hanno portato a mutazioni rilevabili (Veres et al., 2014). Quando si lavora con cloni monocellulari, gli autori notano che “l’eterogeneità clonale può rappresentare un ostacolo più serio alla generazione di linee cellulari veramente isogeniche rispetto agli effetti off-target mediati dai nucleasi”. Inoltre, set di dati su larga scala di centinaia di schermi genetici utilizzando librerie genome-wide hanno mostrato un’elevata concordanza tra diverse sequenze che prendono di mira lo stesso gene, suggerendo che gli effetti off-target non hanno sopraffatto il vero segnale in questi saggi (Dempster et al., 2019). Ancora una volta, la strategia sperimentale è chiara: per qualsiasi gene di interesse, si dovrebbe richiedere che più gRNA di sequenze diverse diano luogo allo stesso fenotipo per concludere che il fenotipo è dovuto a un effetto on-target.
Conclusioni
La selezione dei gRNA per un esperimento deve bilanciare la massimizzazione dell’attività on-target minimizzando l’attività off-target, che sembra ovvio ma spesso può richiedere decisioni difficili. Per esempio, sarebbe meglio usare un gRNA meno attivo che si rivolge a un sito veramente unico nel genoma, o un gRNA più attivo con un sito di destinazione aggiuntivo in una regione del genoma senza funzione nota? Per la creazione di modelli cellulari stabili che devono essere utilizzati per lo studio a lungo termine, il primo può essere la scelta migliore. Per una libreria a livello genomico per condurre schermi genetici, tuttavia, una libreria composta da quest’ultima sarebbe probabilmente più efficace, a condizione che si faccia attenzione nell’interpretazione dei risultati, richiedendo più sequenze che mirano a un gene per ottenere un punteggio al fine di chiamare quel gene un successo.
Questo è un momento eccitante per la genomica funzionale, con una lista sempre crescente di strumenti per sondare la funzione genica. I migliori strumenti sono buoni solo quanto la persona che li usa, e l’uso corretto della tecnologia CRISPR dipenderà sempre da un’attenta progettazione sperimentale, esecuzione e analisi.

Grazie mille al nostro blogger ospite John Doench!
 John Doench è il Direttore di R&D nella Piattaforma di Perturbazione Genetica del Broad Institute e ha lavorato con molti Addgenies per aiutare a migliorare la comprensione, la cura e la spiegazione delle nostre risorse CRISPR. Gli piacciono molto i piccoli RNA.
John Doench è il Direttore di R&D nella Piattaforma di Perturbazione Genetica del Broad Institute e ha lavorato con molti Addgenies per aiutare a migliorare la comprensione, la cura e la spiegazione delle nostre risorse CRISPR. Gli piacciono molto i piccoli RNA.
Anzalone AV, Koblan LW, Liu DR (2020) Editing del genoma con nucleasi CRISPR-Cas, editor di basi, trasposasi e prime editor. Nat Biotechnol 38:824-844 . https://doi.org/10.1038/s41587-020-0561-9
Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H, Allen F, Gonçalves E, Shepherd R, Doench JG, Yusa K, Vazquez F, Parti L, Boehm JS, Golub TR, Hahn WC, Root DE, Garnett MJ, Tsherniak A, Iorio F (2019) Accordo tra due grandi set di dati di dipendenza genica CRISPR-Cas9 pan-cancro. Nat Commun 10: . https://doi.org/10.1038/s41467-019-13805-y
Doench JG (2017) Sono pronto per CRISPR? Una guida dell’utente agli schermi genetici. Nat Rev Genet 19:67-80 . https://doi.org/10.1038/nrg.2017.97
Hanna RE, Doench JG (2020) Progettazione e analisi di esperimenti CRISPR-Cas. Nat Biotechnol 38:813-823 . https://doi.org/10.1038/s41587-020-0490-7
Radzisheuskaya A, Shlyueva D, Müller I, Helin K (2016) L’ottimizzazione della posizione degli sgRNA migliora notevolmente l’efficienza della repressione trascrizionale mediata da CRISPR/dCas9. Nucleic Acids Res 44:e141-e141 . https://doi.org/10.1093/nar/gkw583
Rees HA, Liu DR (2018) Base editing: chimica di precisione sul genoma e trascrittoma delle cellule viventi. Nat Rev Genet 19:770-788 . https://doi.org/10.1038/s41576-018-0059-1
Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K (2014) Bassa incidenza di mutazioni fuori bersaglio in singoli cloni di cellule staminali umane mirate CRISPR-Cas9 e TALEN rilevate da Whole-Genome Sequencing. Cell Stem Cell 15:27-30 . https://doi.org/10.1016/j.stem.2014.04.020
Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Rios X, Huang P-Y, Daley G, Church G (2013) Ottimizzazione del genoma editing delle cellule staminali umane senza cicatrici. Nucleic Acids Research 41:9049-9061 . https://doi.org/10.1093/nar/gkt555
Risorse del Blog Addgene
- Ascolta il nostro Podcast con John Doench
- Impara come condurre schermi di pooled library CRISPR su tutto il genoma
- Leggi altri post del Blog CRISPR
0 commenti