Originally published May 3, 2017 and last updated Sep 24, 2020
Ce billet a été rédigé par le blogueur invité, membre du conseil consultatif d’Addgene et scientifique de l’Institut Broad, John Doench.
La technologieCRISPR a rendu plus facile que jamais à la fois l’ingénierie de modifications spécifiques de l’ADN et la réalisation de cribles fonctionnels pour identifier les gènes impliqués dans un phénotype d’intérêt. Ce billet de blog abordera les différences entre ces approches et fera le point sur la meilleure façon de concevoir des gRNA. Vous pouvez également trouver des gRNA validés pour votre prochaine expérience dans la base de données de séquences de gRNA validés d’Addgene. Une discussion plus approfondie de ces sujets peut être trouvée dans deux articles de synthèse récents (Doench et al., 2017, et Hanna et al., 2020) et leurs références.

Nom nom nom. Me love gRNAs ! Bande dessinée de Maya Kostman.
Considérations importantes avant de commencer une expérience avec CRISPR
Le marteau, la scie sauteuse et la clé à molette sont tous d’excellents outils, mais celui que vous utilisez, bien sûr, dépend de ce que vous essayez de faire – il n’y a pas de » meilleur » outil parmi eux. Bien que cela semble évident, il est important de se rappeler qu’il en va de même lors de la conception d’ARNg pour l’utilisation de la technologie CRISPR – le » meilleur » ARNg dépend énormément de ce que vous essayez de faire : knockout du gène, édition d’une base spécifique ou modulation de l’expression du gène.
 L’emplacement et la séquence sont des considérations importantes pour la conception de vos ARNg. Pour les indels, l’emplacement dans le gène que vous ciblez n’est pas si important, mais il est important que la séquence de votre gRNA soit conçue pour être très active et réduire les cibles. Pour CRISPRa et CRISPRi, ces considérations sont d’une importance à peu près égale (la cible doit être proche du SCT mais vous pouvez moins vous soucier de l’optimalité de la séquence car vous avez généralement moins de séquences parmi lesquelles choisir). Enfin, pour la HDR, la localisation est beaucoup plus importante car vous devez cibler dans un rayon de ~30 nt de votre édition proposée, ce qui signifie qu’il y a si peu de gRNA à choisir que les préférences de séquence doivent largement être ignorées.
L’emplacement et la séquence sont des considérations importantes pour la conception de vos ARNg. Pour les indels, l’emplacement dans le gène que vous ciblez n’est pas si important, mais il est important que la séquence de votre gRNA soit conçue pour être très active et réduire les cibles. Pour CRISPRa et CRISPRi, ces considérations sont d’une importance à peu près égale (la cible doit être proche du SCT mais vous pouvez moins vous soucier de l’optimalité de la séquence car vous avez généralement moins de séquences parmi lesquelles choisir). Enfin, pour la HDR, la localisation est beaucoup plus importante car vous devez cibler dans un rayon de ~30 nt de votre édition proposée, ce qui signifie qu’il y a si peu de gRNA à choisir que les préférences de séquence doivent largement être ignorées.
Le marteau : Assomption de gènes par NHEJ
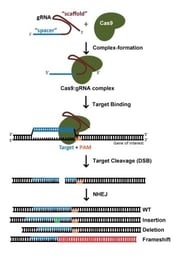 L’assomption de gènes avec la technologie CRISPR est généralement accomplie par des cassures d’ADNdb médiées par Cas9 : après une coupure, la nature sujette aux erreurs de la jonction des extrémités non homologues (NHEJ) conduit souvent à la génération d’indels et donc de décalages de cadre qui perturbent la capacité de codage des protéines d’un locus. Lorsque l’on utilise S. pyogenes Cas9 (SpCas9), les sites cibles potentiels sont à la fois et , car il est tout aussi efficace de cibler le brin codant ou non codant de l’ADN. En règle générale, nous évitons les sites cibles qui codent pour des acides aminés proches de l’extrémité N’ de la protéine, afin d’atténuer la capacité de la cellule à utiliser un ATG alternatif en aval du codon de départ annoté. De même, nous évitons les sites cibles qui codent pour des acides aminés proches de l’extrémité C’ de la protéine, afin de maximiser les chances de créer un allèle non fonctionnel. Pour un gène d’un kilobase, étant donné que les sites cibles potentiels sont présents à raison d’un nucléotide sur huit, le fait de limiter les ARNg à 5 à 65 % de la région codant pour la protéine permettra de choisir parmi plusieurs dizaines d’ARNg. Avec autant de possibilités, choisir un gRNA avec une séquence optimisée est d’une importance primordiale (plus sur ce point ci-dessous).
L’assomption de gènes avec la technologie CRISPR est généralement accomplie par des cassures d’ADNdb médiées par Cas9 : après une coupure, la nature sujette aux erreurs de la jonction des extrémités non homologues (NHEJ) conduit souvent à la génération d’indels et donc de décalages de cadre qui perturbent la capacité de codage des protéines d’un locus. Lorsque l’on utilise S. pyogenes Cas9 (SpCas9), les sites cibles potentiels sont à la fois et , car il est tout aussi efficace de cibler le brin codant ou non codant de l’ADN. En règle générale, nous évitons les sites cibles qui codent pour des acides aminés proches de l’extrémité N’ de la protéine, afin d’atténuer la capacité de la cellule à utiliser un ATG alternatif en aval du codon de départ annoté. De même, nous évitons les sites cibles qui codent pour des acides aminés proches de l’extrémité C’ de la protéine, afin de maximiser les chances de créer un allèle non fonctionnel. Pour un gène d’un kilobase, étant donné que les sites cibles potentiels sont présents à raison d’un nucléotide sur huit, le fait de limiter les ARNg à 5 à 65 % de la région codant pour la protéine permettra de choisir parmi plusieurs dizaines d’ARNg. Avec autant de possibilités, choisir un gRNA avec une séquence optimisée est d’une importance primordiale (plus sur ce point ci-dessous).
Le puzzle : Édition par HDR, édition des bases et édition primaire
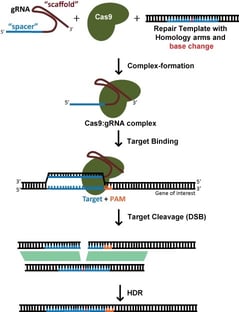
Pour une édition spécifique, comme l’insertion d’une étiquette fluorescente ou l’introduction d’une mutation spécifique, on s’appuie généralement sur la réparation dirigée par l’homologie (HDR) pour incorporer de nouvelles informations dans l’ADN. Cela nécessite également un modèle d’ADN exogène. Cependant, la réparation dirigée par l’homologie est un processus à très faible efficacité, qui nécessite généralement le clonage d’une seule cellule et le dépistage ultérieur des modifications réussies. Ce processus prend beaucoup de temps et ne doit pas être entrepris à la légère ! En effet, atteindre véritablement l’étalon-or nécessite non pas une mais deux séries de clonage de cellules uniques – à titre de contrôle, il faudrait revenir à l’édition d’origine afin de prouver que le phénotype était vraiment dû à l’édition prévue plutôt qu’à une variante passagère qui est arrivée avec le clone de cellule unique (bien que cela soit rarement fait).
Lorsqu’on cible une cassure d’ADNdb pour la HDR, le choix du site cible est beaucoup plus contraint par la localisation souhaitée de l’édition ; l’efficacité diminue considérablement lorsque le site de coupe est >30nt des extrémités proximales du modèle de réparation (Yang et al, 2013). Cela signifie que, pour l’édition de gènes, il y a généralement peu de gRNAs potentiels. Bien que SpCas9, avec une préférence PAM de NGG, soit encore l’enzyme Cas la plus utilisée, le développement des enzymes SaCas9, NmeCas9, Cas12a et de leurs variantes modifiées offre des options PAM supplémentaires qui peuvent élargir considérablement les options gRNA.
Deux technologies plus récentes offrent une alternative à la HDR pour introduire des éditions. Les mêmes contraintes de localisation sont encore plus exquises pour le soi-disant éditeur de bases Cas9, qui effectue des modifications de l’ADN en l’absence de cassures d’ADNdb (Rees et al., 2018). Pour les éditeurs de bases C>T et A>G, l’édition prévue doit se trouver dans une fenêtre de 5 à 10 nt par rapport au PAM, et les éditions indirectes sont possibles s’il y a un autre C ou A cible dans la fenêtre. Une autre technologie, l’édition de prime (revue dans Anzalone et al., 2020). n’est pas limitée aux transitions de nucléotides uniques mais nécessite toujours un PAM à proximité, bien que ce soit encore les premiers jours pour cette technologie, et l’utilisateur peut avoir besoin d’optimiser de nombreux paramètres pour générer l’édition souhaitée.
La clé : L’activation et l’inhibition des gènes par CRISPRa et CRISPRi
Enfin, pour moduler l’expression des gènes au niveau de la transcription – technologies CRISPRa (activation) et CRISPRi (inhibition) – un Cas9 (dCas9) sans nucléase est dirigé près du promoteur d’un gène cible. Ici, la fenêtre cible n’est pas aussi large que pour le knockout par coupure CRISPR. Pour CRISPRa, il est plus efficace de cibler une fenêtre de ~100nt en amont du site de début de transcription (SCT), tandis que pour CRISPRi, une fenêtre de ~100nt en aval du SCT donne le plus d’activité. Ainsi, pour un gène donné, il n’y aura qu’une douzaine d’ARNg à choisir dans l’emplacement optimal. Il est également important d’avoir de bonnes informations sur l’emplacement exact du SCT. Différentes bases de données annotent le SCT de différentes manières, et il a été démontré que la base de données FANTOM, qui s’appuie sur CAGE-seq pour capturer directement la coiffe de l’ARNm, fournit la cartographie la plus précise (Radzisheuskaya et al., 2016). Dans ce cas, l’emplacement et la séquence ont une importance à peu près égale dans la conception – une séquence optimisée ne fera pas grand-chose si elle est au mauvais endroit, mais comme la fenêtre cible est plus étroite, il y a moins de gRNA parmi lesquels choisir, et donc une séquence optimale peut ne pas être disponible.
Prédire l’efficacité des gRNA
Nous et d’autres avons examiné la possibilité d’utiliser des caractéristiques basées sur la séquence et d’autres caractéristiques pour nommer les gRNA qui sont susceptibles d’être actifs, non seulement pour SpCas9 mais aussi pour certaines autres enzymes Cas. Il semble qu’il n’existe pas de système de notation universel pour sélectionner un gRNA, car la méthode de production du guide (synthétique, transcription in vitro ou livraison lentivirale) peut affecter la précision d’un score prédictif, ainsi que les aspects dynamiques de la cible (par exemple, l’accessibilité due à l’état de la chromatine). Aucune prédiction computationnelle n’est jamais parfaite, mais cela peut diminuer le nombre de guides que l’on doit tester en laboratoire.
Important, pour toute modification d’intérêt, il serait imprudent de tirer des conclusions sur la base de l’activité d’un seul gRNA, et donc la diversité des gRNA à travers un gène devrait être examinée chaque fois que possible lors de l’utilisation d’approches de knockout ou de modulation transcriptionnelle.
Éviter les effets hors cible

L’activité hors cible des gRNA est importante à considérer. Si le paysage de base des mésappariements qui peuvent néanmoins conduire à une activité a été établi, et peut être utilisé pour identifier les sites susceptibles de donner lieu à un effet hors cible, il n’y a pas suffisamment de données pour prédire pleinement quels sites présenteront ou non des niveaux appréciables de modification. Le séquençage du génome entier de cellules modifiées par CRISPR indique que les conséquences de l’activité hors cible, du moins pour les conditions expérimentales utilisées, n’ont entraîné aucune mutation détectable (Veres et al., 2014). Lorsqu’on travaille avec des clones unicellulaires, les auteurs notent que « l’hétérogénéité clonale peut représenter un obstacle plus sérieux à la génération de lignées cellulaires véritablement isogéniques que les effets hors cible médiés par les nucléases. » En outre, les ensembles de données à grande échelle de centaines de cribles génétiques utilisant des bibliothèques pangénomiques ont montré une concordance élevée entre différentes séquences ciblant le même gène, ce qui suggère que les effets hors cible n’ont pas écrasé le véritable signal dans ces essais (Dempster et al., 2019). Là encore, la stratégie expérimentale est claire : pour tout gène d’intérêt, il faut exiger que plusieurs ARNg de séquences différentes donnent lieu au même phénotype pour conclure que le phénotype est dû à un effet on-target.
Conclusions
La sélection des ARNg pour une expérience doit trouver un équilibre entre la maximisation de l’activité on-target et la minimisation de l’activité off-target, ce qui semble évident mais peut souvent nécessiter des décisions difficiles. Par exemple, serait-il préférable d’utiliser un ARNg moins actif qui cible un site vraiment unique dans le génome, ou un ARNg plus actif avec un site cible supplémentaire dans une région du génome sans fonction connue ? Pour la création de modèles cellulaires stables destinés à être utilisés pour des études à long terme, le premier choix peut être le meilleur. En revanche, pour une bibliothèque à l’échelle du génome destinée à réaliser des cribles génétiques, une bibliothèque composée des seconds serait probablement plus efficace, tant que l’on prend soin d’interpréter les résultats en exigeant que plusieurs séquences ciblant un gène soient marquées pour que ce gène soit qualifié de hit.
C’est une période passionnante pour la génomique fonctionnelle, avec une liste toujours plus longue d’outils pour sonder la fonction des gènes. Les meilleurs outils ne sont aussi bons que la personne qui les utilise, et la bonne utilisation de la technologie CRISPR dépendra toujours d’une conception, d’une exécution et d’une analyse expérimentales minutieuses.

Mon grand merci à notre blogueur invité John Doench !
 John Doench est le directeur de R&D de la plateforme de perturbation génétique au Broad Institute et a travaillé avec de nombreux Addgenies pour aider à améliorer la compréhension, la curation et l’explication de nos ressources CRISPR. Il aime beaucoup les petits ARN.
John Doench est le directeur de R&D de la plateforme de perturbation génétique au Broad Institute et a travaillé avec de nombreux Addgenies pour aider à améliorer la compréhension, la curation et l’explication de nos ressources CRISPR. Il aime beaucoup les petits ARN.
Anzalone AV, Koblan LW, Liu DR (2020) Édition du génome avec les nucléases CRISPR-Cas, les éditeurs de bases, les transposases et les éditeurs de prime. Nat Biotechnol 38:824-844 . https://doi.org/10.1038/s41587-020-0561-9
Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H, Allen F, Gonçalves E, Shepherd R, Doench JG, Yusa K, Vazquez F, Parts L, Boehm JS, Golub TR, Hahn WC, Root DE, Garnett MJ, Tsherniak A, Iorio F (2019) Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat Commun 10 : . https://doi.org/10.1038/s41467-019-13805-y
Doench JG (2017) Am I ready for CRISPR ? Un guide de l’utilisateur pour les écrans génétiques. Nat Rev Genet 19:67-80 . https://doi.org/10.1038/nrg.2017.97
Hanna RE, Doench JG (2020) Conception et analyse des expériences CRISPR-Cas. Nat Biotechnol 38:813-823 . https://doi.org/10.1038/s41587-020-0490-7
Radzisheuskaya A, Shlyueva D, Müller I, Helin K (2016) L’optimisation de la position des sgRNA améliore nettement l’efficacité de la répression transcriptionnelle médiée par CRISPR/dCas9. Nucleic Acids Res 44:e141-e141 . https://doi.org/10.1093/nar/gkw583
Rees HA, Liu DR (2018) Base editing : precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19:770-788 . https://doi.org/10.1038/s41576-018-0059-1
Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K (2014) Faible incidence des mutations hors cible dans les clones individuels de cellules souches humaines ciblés par CRISPR-Cas9 et TALEN détectés par séquençage du génome entier. Cell Stem Cell 15:27-30 . https://doi.org/10.1016/j.stem.2014.04.020
Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Rios X, Huang P-Y, Daley G, Church G (2013) Optimisation de l’édition du génome de cellules souches humaines sans cicatrices. Nucleic Acids Research 41:9049-9061 . https://doi.org/10.1093/nar/gkt555
Ressources sur le blog Addgene
- Écouter notre podcast avec John Doench
- Apprendre comment réaliser des cribles de bibliothèques groupées CRISPR à l’échelle du génome
- Lire d’autres articles du blog CRISPR
.
0 commentaire