Introducción
El rabdomiosarcoma (RMS) es un tumor maligno del músculo estriado de origen mesenquimal considerado como el sarcoma de tejidos blandos más recurrente en niños y adolescentes, con una incidencia anual de 4,3 casos por millón. Aproximadamente dos tercios de los casos se diagnostican en niños (normalmente en torno a los 6 años de edad), y son ligeramente más frecuentes en los varones, con una proporción hombre/mujer de 1,4:1.1 Debido a su origen en una célula totipotente, los rabdomiosarcomas no sólo se producen en el músculo esquelético sino en otras localizaciones, como la cabeza, el cuello, el tracto genitourinario y los conductos biliares. Aproximadamente el 40% de los RMS se producen en la región de la cabeza y el cuello, el 20% en la región genitourinaria, el 20% en las extremidades y el 20% restante en otras localizaciones.1
Las variantes embrionarias y alveolares son los tipos histológicos más frecuentes, comprendiendo del 70 al 20% de los casos, respectivamente.2 El rabdomiosarcoma embrionario (RMS) es el subtipo más frecuente en lactantes y niños pequeños y representa más de dos tercios de todos los RMS.1,3 Este tumor está compuesto por una mezcla blanda de células fusiformes con áreas de estroma blando y suelto, y se asocia a la pérdida de heterocigosidad en el locus 11p15.3 Los tumores que se localizan en la cabeza, el cuello y la región genitourinaria suelen presentar este tipo histológico.
Los rabdomiosarcomas alveolares (ARMS) suelen aparecer en la adolescencia; se localizan típicamente en las extremidades y tienen una alta capacidad de metástasis.1,4 Su histología se caracteriza por un tabique de tejido conectivo fibroso con células neoplásicas adheridas (Figura 1A), similar a los espacios alveolares observados en el pulmón, donde algunas de las células se desprenden y ocupan el espacio. Está compuesto por células uniformemente poligonales con núcleo redondo u ovalado hipercromático de alto grado (Figura 1B).1 A diferencia de los ERMS, los ARMS presentan dos tipos particulares de translocaciones cromosómicas: entre los cromosomas 2 y 13, t (2;13) (q35;q14), y entre los cromosomas 1 y 13, t (1;13) (p36;q14), que se dan en el 80% de los casos.4 Estas alteraciones genéticas conducen a la fusión de dos familias de factores de transcripción. La primera se localiza en el cromosoma 1 o 2 y afecta a los factores de transcripción PAX3 y PAX7, respectivamente. La familia de factores de transcripción PAX es un grupo de genes implicados en la diferenciación de órganos y tejidos, y poseen un dominio de unión al ADN N-terminal, que incluye una caja emparejada, motivos homeobox, y un dominio de trans-activación C-terminal, mientras que la segunda clase implica a miembros de la familia de los factores de transcripción forkhead (FKHR) o FOXO1.
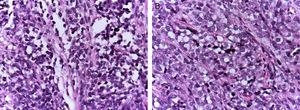
(A) Aspecto histológico del rabdomiosarcoma alveolar: los septos conectivos fibrosos forman estructuras pseudoalveolares, en las que se incrustan las células neoplásicas. (B) En algunas zonas, ocupan todo el espacio formando una neoplasia sólida.
Los factores de transcripción de las familias PAX y FOXO1 poseen un dominio N-terminal de unión con el ADN y un dominio C-terminal de transactivación. Los puntos de ruptura de PAX y FOXO1 se producen en los intrones 7 y 1, respectivamente. Los genes fusionados codifican dos proteínas quiméricas con actividad oncogénica, PAX3/FOXO1, y PAX7/FOXO1. Estas proteínas están compuestas por un dominio 5′ de unión al ADN (PAX) y un dominio 3′ de transactivación (FOXO1).4 Se ha demostrado que los transcritos PAX3/FOXO1 y PAX7/FOXO1 están presentes en el 55 y el 22% de los ARMS, respectivamente, mientras que los ARMS restantes son negativos para el gen de fusión.5 Se sabe que los ARMS con la translocación PAX7/FOXO1 tienen un pronóstico mucho más favorable en comparación con los que llevan la translocación PAX3/FOXO1. La mediana de supervivencia a los 4 años para los primeros es del 75% y del 8% para los segundos.6 Los pacientes con ARMS que suelen tener metástasis en el momento del diagnóstico tienen una mediana de supervivencia corta. Además, la presencia de proteínas del gen de fusión PAX3/7-FOXO1 se asocia con un pronóstico desfavorable.2
Caso clínico
Un paciente varón de dos años y tres meses de edad, originario de Yucatán, que no tenía antecedentes médicos significativos hasta los cinco meses de edad, cuando apareció un bulto violeta en su fosa nasal izquierda. Según los padres, fue tratado con quimioterapia y radioterapia en una clínica local, con remisión del tumor dos meses después. Cuando el niño tenía dos años, el tumor volvió a aparecer, por lo que acudieron a nuestro hospital.
En la exploración física, su peso era de 10,4 kg y su altura de 100 cm. Estaba despierto, su ojo derecho tenía reflejo pupilar luminoso normal, pero el ojo izquierdo no era evaluable debido a un tumor localizado en la mitad de la cara, predominantemente en el lado izquierdo, violáceo, fétido, fijado a planos profundos. Los dientes estaban desplazados. No tenía linfadenopatías en el cuello. No se encontraron anomalías en el tórax, el abdomen o las extremidades.
La tomografía computarizada (TC) mostró un tumor lobulado, de bordes bien definidos que comprometía desde la región frontal hasta el piso maxilar. El diagnóstico histopatológico fue de rabdomiosarcoma alveolar en estadio IV con infiltración a médula ósea y líquido cefalorraquídeo. Se trató con antibióticos y se inició la quimioterapia con adriamicina, actinomicina, ciclofosfamida y vincristina. Se completó el primer ciclo y fue dado de alta. Recibió cuatro ciclos de quimioterapia, con una reducción del tamaño del tumor del 50%. En el último ciclo, se añadió cisplatino e irinotecán al régimen quimioterapéutico. El último TAC mostró ensanchamiento de la cavidad nasal y del antro maxilar con deformidad del lado izquierdo de la cara, desplazamiento del globo ocular, infiltración hasta la pared medial de la órbita, cornetes mal definidos, paredes medial y lateral del antro maxilar izquierdo y ocupación de los senos esfenoidales.
Se suspendió el cuarto ciclo quimioterapéutico por fiebre y neutropenia, distensión abdominal y disminución del peristaltismo por hipopotasemia e íleo metabólico. En el hemocultivo se aisló un bacilo gramnegativo. Se colocó una sonda nasogástrica y se descartó una colitis neutropénica. La insuficiencia respiratoria llevó a la ventilación mecánica invasiva. El paciente presentaba desequilibrio electrolítico y shock séptico refractario, y finalmente falleció.
Discusión
El rabdomiosarcoma es el sarcoma de tejidos blandos más frecuente en niños; se clasifica en rabdomiosarcoma embrionario (ERMS), rabdomiosarcoma alveolar (ARMS), rabdomiosarcoma botrioide y rabdomiosarcoma de células fusiformes, con diferentes fenotipos y características clínicas (figura 2). Recientemente se han descrito variantes menos comunes, como el rabdomiosarcoma esclerosante y los de características rabdoides. El ERMS y el ARMS son los más prevalentes y comprenden el 70% y el 20% de los casos, respectivamente. De ellos, el ARMS es el de peor pronóstico.2,3 Este comportamiento se ha asociado a la expresión de proteínas de fusión oncogénicas resultantes de translocaciones cromosómicas, un mecanismo de tumorigénesis común con muchos tipos de cáncer, incluyendo un tercio de los sarcomas.5
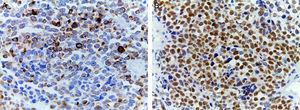
(A) Tinción inmunohistoquímica que muestra positividad para desmina. En el citoplasma de las células musculares estriadas está presente un filamento intermedio asociado a bandas Z. (B) Positividad nuclear intensa para la mioglobina, una hemoproteína presente en el músculo esquelético y que sirve como transportador y reservorio de oxígeno; aparece tarde en la maduración secuencial del músculo y es positiva en el 95% de los casos de rabdomiosarcomas.
La translocación cromosómica más común encontrada en los ARMS es t(2;13) (q35;q14) PAX3/FOXO1, y la menos frecuente es t(1;13) (p36;q14) PAX7/FOXO1.6,7 Las translocaciones t(2;13) y t(1;13) son el resultado de la ruptura de genes específicos que se encuentran dentro de la región cromosómica 2q35 y 1p36, respectivamente, seguida de una fusión. Los genes implicados en el cromosoma 2 y 1 son PAX3 y PAX7, respectivamente, que codifican para factores de transcripción de la familia de cajas pareadas.3 Los genes que se fusionan con PAX3 y PAX7 son FOXO1 o FKHR, que se encuentran en el cromosoma 13 y son miembros de la familia de factores de transcripción forkhead. La fusión de estos genes da lugar a la expresión de proteínas de fusión, que actúan como activadores transcripcionales que contribuyen al desarrollo tumoral alterando las vías de crecimiento celular y apoptosis, modulando la diferenciación miogénica y estimulando la motilidad y otras vías metastásicas.3,8 Durante el desarrollo normal, la expresión de PAX3 es necesaria para la migración de los precursores de las células musculares esqueléticas hacia las extremidades, mientras que la expresión de PAX7, un marcador de las células satélite del músculo esquelético en los adultos, es necesaria para el recambio celular normal. Ambas proteínas se degradan rápidamente durante la diferenciación miogénica temprana. Sin embargo, en el ARMS, la fusión de PAX3/PAX7 con FOXO1 les confiere una vida media más larga.7
La RT-PCR y la hibridación fluorescente in situ son métodos que se han utilizado para analizar la frecuencia de la fusión de PAX3 y PAX7 en el ARMS (Figura 3). El Intergroup Rhabdomyosarcoma Study (IRS), que incluyó 78 casos de ARMS, encontró que el 77% de los casos eran positivos para la fusión, el 55% de los ARMS expresaban PAX3/FOXO1, el 22% expresaban PAX7/FOXO1, y el 23% eran negativos para la fusión.9 En un estudio publicado en 2010, encontramos una tendencia similar: en 25 casos de ARMS encontramos que el 50% eran positivos para PAX3/FOXO1, el 3% eran positivos para PAX7/FOXO1, y el 30% eran negativos para el gen de fusión.10 En este estudio, se analizaron 37 casos de ARMS, de los cuales el 63% eran positivos para PAX3/FOXO1, el 5% eran positivos para PAX7/FOXO1, y el 32% eran negativos para la fusión, lo que coincide con otros informes. La detección de las proteínas de fusión oncogénicas, en particular PAX3/FOXO1, tiene un valor pronóstico significativo, ya que el comportamiento biológico del tumor en el RMS con esta translocación es más agresivo, con mala respuesta al tratamiento y frecuentes recidivas. Por el contrario, los tumores con la translocación PAX7/FOXO1 tienen una mejor respuesta al tratamiento, un estadio clínico generalmente más bajo y una mayor supervivencia.11
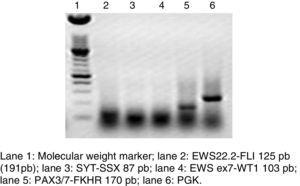
Electroforesis en gel de agarosa de una RT-PCR, donde se muestra la translocación PAX3/7-FKHR representativa del rabdomiosarcoma alveolar, descartando otras neoplasias sólidas.
Los mecanismos por los que la proteína quimérica PAX/FOXO1 contribuye a la oncogénesis del RMS han sido profundamente estudiados. Aparentemente, la cantidad y la localización celular de la proteína son críticas para su actividad oncogénica. Tanto PAX3/FOXO1 como PAX7/FOXO1 tienen una actividad transcripcional 100 veces mayor que las proteínas silvestres PAX3 y PAX7.8,9 Las proteínas PAX/FOXO1 se expresan por sí solas a altos niveles. La sobreexpresión de PAX3 resulta de un aumento de la transcripción que es independiente del número de copias, mientras que la alta expresión de PAX7 se asocia a la amplificación del gen.12,13 Aparte de su sobreexpresión, PAX3/FOXO1 es significativamente más estable que PAX3, que sufre rápidamente la proteólisis durante la diferenciación muscular.14 Dentro de la célula, estas proteínas quiméricas pueden encontrarse en el núcleo o en el citoplasma. En condiciones normales, la localización de la FOXO1 salvaje está controlada por AKT. Cuando AKT es estimulada, fosforila FOXO1, provocando su retención en el citoplasma. Sin embargo, en el ARMS, la proteína de fusión PAX/FOXO1 es resistente a la actividad de AKT y permanece predominantemente en el núcleo.15 Otra forma en la que la proteína PAX/FOXO1 contribuye en la oncogénesis es impidiendo la apoptosis de la célula tumoral a través de la expresión de genes antiapoptóticos como Bcl-XL.
El principal problema en la biología de los rabdomiosarcomas es su susceptibilidad a diferenciarse en músculo esquelético de forma indefinida, lo cual es el resultado de alteraciones en el programa miogénico, a nivel de quinasas (por ejemplo, p38 MAPK) y epigenético. Dentro de este último, se encuentran el complejo policombrepresor o JARID2, y los microRNAs (miRNAs).16 Los miRNAs son una familia de RNAs no codificantes que regulan la expresión génica a nivel post-transcripcional mediante la inhibición o degradación del RNA mensajero. En la actualidad, se sabe que hay un número importante de miARNs implicados en la inducción y progresión de los rabdomiosarcomas. La expresión de varios miRNAs se induce durante el proceso miogénico, y sus potenciales dianas son genes que controlan la proliferación y diferenciación de los mioblastos, dando lugar a una desregulación de la proliferación celular o a una diferenciación miogénica aberrante. Los miRNAs más estudiados en el músculo esquelético son miRNA-1, miRNA-133a/b, y miRNA-206, que son específicos del músculo, y miRNA-29b/c que se expresa de forma ubicua.17
Existen evidencias que apuntan a la familia de miRNA-29 como supresores de tumores ya que se ha observado la expresión aberrante de miembros de esta familia en varios tipos de cáncer. La familia miRNA-29s es una familia conservada, que incluye miRNA-29a/b/c, que tiene un papel importante en la proliferación celular, la apoptosis, la migración y la invasión.17-19 miRNA-29b es el miembro de esta familia que se expresa en niveles más altos en condiciones normales.19
El pronóstico de los pacientes con RMS depende del grado del tumor, de la edad, del tipo de resección, de la histología, de la presencia de las translocaciones mencionadas y del número de focos con metástasis.
La transición epitelio-mesénquima (EMT) es un evento importante para la invasión y metástasis del tumor, y se asocia con un mal pronóstico y con la quimiorresistencia. La EMT es un proceso en el que las células epiteliales pierden su polaridad y la adhesión célula-célula, y pueden migrar e invadir otros órganos.20 Este proceso se inicia con la disociación de las uniones intercelulares (claudina, ocludina, ZO-1, E-cadherina y desmoplakina), dando lugar a la pérdida de las microvellosidades y de la polaridad apical-basolateral.
Las células adquieren una morfología alargada, aumentan la expresión de α-actina de músculo liso y potencian su capacidad de migración. En la última etapa de la EMT, las células adquieren el potencial de degradar la membrana basal a través de la expresión de metaloproteinasas de matriz (MMP).20 La EMT se acompaña de cambios moleculares como la baja expresión de citoqueratina y vimentina, la sobreexpresión de N-cadherina y el factor de transcripción Snail (inhibidor de la expresión de E-cadherina).20
La quimiorresistencia es uno de los principales problemas en casi todos los tipos de cáncer. A pesar de que hay una mejora en los agentes quimioterapéuticos, muchos pacientes con cáncer mueren porque desarrollan quimiorresistencia. Varios estudios indican que los miRNAs están implicados en la quimiorresistencia,21 entre ellos el miRNA-29b.
El tipo de quimioterapia que reciben los pacientes con RMS depende de los factores de riesgo que presenten. Los pacientes de riesgo bajo o intermedio reciben vincristina, dactinomicina y ciclofosfamida.22,23 En algunos pacientes de riesgo intermedio, la intensificación de la dosis de ciclofosfamida tras la resección total da buenos resultados; sin embargo, en otros pacientes, la intensificación de la quimioterapia no mejora el resultado.24 En los pacientes de alto riesgo, se utiliza la combinación de ifosfamida-etopósido o ifosfamida y doxorrubicina. Estos pacientes suelen tener un mal pronóstico. Curiosamente, se ha observado una buena respuesta al tratamiento en pacientes con EMR de alto riesgo con una o más metástasis. Este tipo de RMS tiene un mejor pronóstico que otros RMS metastásicos.25 El ARMS de fusión negativa se comporta de forma similar al ERMS.7 Hemos encontrado que la expresión de miRNA-29b es mayor en los tumores con fusión negativa, y tienen una mejor respuesta a la quimioterapia.
Divulgación éticaProtección de los sujetos humanos y animales
Los autores declaran que los procedimientos seguidos se ajustaron a las normas del comité de ética de investigación clínica correspondiente y a las del Código de Ética de la Asociación Médica Mundial (Declaración de Helsinki).
Confidencialidad de los datos
Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la intimidad y consentimiento informado.
0 comentarios