Introduction
Le rhabdomyosarcome (RMS) est une tumeur maligne du muscle strié d’origine mésenchymateuse considérée comme le sarcome des tissus mous le plus récurrent chez les enfants et les adolescents, avec une incidence annuelle de 4,3 cas par million. Environ deux tiers des cas sont diagnostiqués chez les enfants (généralement vers l’âge de 6 ans), et sont légèrement plus fréquents chez les garçons, avec un rapport homme/femme de 1,4:1.1 En raison de son origine dans une cellule totipotente, les rhabdomyosarcomes ne se produisent pas seulement dans les muscles squelettiques, mais aussi dans d’autres endroits, comme la tête, le cou, les voies génito-urinaires et les canaux biliaires. Environ 40 % des RMS surviennent dans la région de la tête et du cou, 20 % dans la région génito-urinaire, 20 % dans les extrémités et les 20 % restants dans d’autres sites.1
Les variantes embryonnaires et alvéolaires sont les types histologiques les plus fréquents, comprenant respectivement 70 à 20 % des cas.2
Les rhabdomyosarcomes embryonnaires sont les plus fréquents. Le rhabdomyosarcome embryonnaire (ERMS) est le sous-type le plus fréquent chez les nourrissons et les jeunes enfants et représente plus de deux tiers de tous les RMS.1,3 Cette tumeur est composée d’un mélange mou de cellules fusiformes avec des zones de stroma mou et lâche, et elle est associée à une perte d’hétérozygotie au locus 11p15.3 Les tumeurs qui sont situées dans la tête, le cou et la région génito-urinaire présentent souvent ce type histologique.
Les rhabdomyosarcomes alvéolaires (RMA) apparaissent généralement à l’adolescence ; ils sont typiquement situés dans les extrémités et ont une grande capacité à métastaser1,4. Leur histologie est caractérisée par un septum de tissu conjonctif fibreux auquel sont attachées des cellules néoplasiques (figure 1A), semblable aux espaces alvéolaires observés dans le poumon, où certaines des cellules se détachent et occupent l’espace. Il est composé de cellules uniformément polygonales avec un noyau hyperchromatique rond ou ovale de haut grade (Figure 1B).1 Contrairement aux ERMS, les ARMS présentent deux types particuliers de translocations chromosomiques : entre les chromosomes 2 et 13, t (2;13) (q35;q14), et entre les chromosomes 1 et 13, t (1;13) (p36;q14), qui se produisent dans 80 % des cas.4 Ces altérations génétiques entraînent la fusion de deux familles de facteurs de transcription. La première est située sur le chromosome 1 ou 2 et implique les facteurs de transcription PAX3 et PAX7, respectivement. La famille des facteurs de transcription PAX est un groupe de gènes impliqués dans la différenciation des organes et des tissus, et possèdent un domaine de liaison à l’ADN N-terminal, qui comprend une boîte appariée, des motifs homéobox, et un domaine de trans-activation C-terminal, tandis que la seconde classe implique les membres de la famille des facteurs de transcription forkhead (FKHR) ou FOXO1.
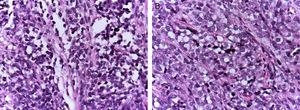
(A) Aspect histologique d’un rhabdomyosarcome alvéolaire : des septa conjonctifs fibreux forment des structures pseudo-alvéolaires, dans lesquelles sont incluses des cellules néoplasiques. (B) Dans certaines zones, elles occupent tout l’espace formant un néoplasme solide.
Les facteurs de transcription des familles PAX et FOXO1 possèdent un domaine N-Terminal d’union avec l’ADN et un domaine C-terminal de transactivation. Les points de rupture pour PAX et FOXO1 se situent respectivement au niveau des introns 7 et 1. Les gènes fusionnés codent pour deux protéines chimériques à activité oncogène, PAX3/FOXO1, et PAX7/FOXO1. Ces protéines sont composées d’un domaine 5′ d’union à l’ADN (PAX) et d’un domaine 3′ de transactivation (FOXO1).4 Il a été démontré que les transcrits PAX3/FOXO1 et PAX7/FOXO1 sont présents dans 55 et 22% des SMRA, respectivement, tandis que les SMRA restants sont négatifs pour le gène de fusion.5 Il est connu que les SMRA avec la translocation PAX7/FOXO1 ont un pronostic beaucoup plus favorable par rapport à ceux qui portent la translocation PAX3/FOXO1. La survie médiane à 4 ans est de 75% pour les premiers et de 8% pour les seconds.6 Les patients atteints de SRA qui présentent souvent des métastases au moment du diagnostic ont une survie médiane courte. De plus, la présence de protéines du gène de fusion PAX3/7-FOXO1 est associée à un pronostic défavorable.2
Cas clinique
Un patient de sexe masculin âgé de deux ans et trois mois, originaire du Yucatan, qui n’avait pas d’antécédents médicaux significatifs jusqu’à l’âge de cinq mois où un bouchon violet est apparu dans sa narine gauche. Selon les parents, il a été traité par chimiothérapie et radiothérapie dans une clinique locale, avec une rémission de la tumeur deux mois plus tard. Lorsque le garçon avait deux ans, la tumeur est réapparue, alors ils sont venus à notre hôpital.
À l’examen physique, son poids était de 10,4 kg et sa taille de 100 cm. Il était éveillé, son œil droit avait un réflexe pupillaire lumineux normal, mais l’œil gauche n’était pas évaluable en raison d’une tumeur située au milieu du visage, prédominant sur le côté gauche, violacée, fétide, fixée aux plans profonds. Les dents étaient déplacées. Il n’avait pas d’adénopathies au niveau du cou. Aucune anomalie n’était retrouvée au niveau du thorax, de l’abdomen ou des membres.
La tomodensitométrie (TDM) a montré une tumeur lobée, aux limites bien définies qui impliquait la région frontale jusqu’au plancher maxillaire. Le diagnostic histopathologique était un rhabdomyosarcome alvéolaire de stade IV avec une infiltration de la moelle osseuse et du liquide céphalo-rachidien. Il a été traité par antibiotiques et a commencé une chimiothérapie à base d’adriamycine, d’actinomycine, de cyclophosphamide et de vincristine. Le premier cycle a été achevé et il est sorti de l’hôpital. Il a reçu quatre cycles de chimiothérapie, avec une réduction de la taille de la tumeur de 50 %. Au cours du dernier cycle, le cisplatine et l’irinotécan ont été ajoutés au régime chimiothérapeutique. Le dernier scanner a montré un élargissement de la cavité nasale et de l’antre maxillaire avec déformation du côté gauche du visage, un déplacement du globe oculaire, une infiltration jusqu’à la paroi médiale de l’orbite, des turbinats mal définis, des parois médiale et latérale de l’antre maxillaire gauche et une occupation des sinus sphénoïdes.
Le quatrième cycle de chimiothérapie a été suspendu en raison de la fièvre et de la neutropénie, du ballonnement abdominal et de la diminution du péristaltisme en raison de l’hypokaliémie et de l’iléus métabolique. Un bacille gram-négatif a été isolé dans l’hémoculture. Une sonde nasogastrique a été placée, et la colite neutropénique a été écartée. L’insuffisance respiratoire a conduit à une ventilation mécanique invasive. Le patient présentait un déséquilibre électrolytique et un choc septique réfractaire, et est finalement décédé.
Discussion
Le rhabdomyosarcome est le sarcome des tissus mous le plus fréquent chez les enfants ; il est classé en rhabdomyosarcome embryonnaire (ERMS), rhabdomyosarcome alvéolaire (ARMS), rhabdomyosarcome botryoïde et rhabdomyosarcome à cellules fusiformes, avec des phénotypes et des caractéristiques cliniques différents (figure 2). Des variantes moins courantes ont été récemment décrites, comme les rhabdomyosarcomes sclérosés et ceux présentant des caractéristiques rhabdoïdes. Les ERMS et ARMS sont les plus répandus et représentent respectivement 70% et 20% des cas. Parmi ceux-ci, l’ARMS est celui dont le pronostic est le plus mauvais.2,3 Ce comportement a été associé à l’expression de protéines de fusion oncogènes résultant de translocations chromosomiques, un mécanisme de tumorigenèse commun à de nombreux types de cancers, dont un tiers des sarcomes.5
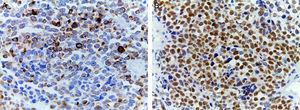
(A) Coloration immunohistochimique montrant la positivité de la desmine. Un filament intermédiaire est présent dans le cytoplasme des cellules musculaires striées associé à des bandes Z. (B) Intense positivité nucléaire pour la myoglobine, une hémoprotéine présente dans le muscle squelettique et servant de transporteur et de réservoir d’oxygène ; elle apparaît tardivement sur la maturation musculaire séquentielle et est positive dans 95% des cas de rhabdomyosarcomes.
La translocation chromosomique la plus fréquemment retrouvée dans les SRA est la t(2;13) (q35;q14) PAX3/FOXO1, et la moins fréquemment retrouvée est la t(1;13) (p36;q14) PAX7/FOXO1.6,7 Les translocations t(2;13) et t(1;13) résultent de la rupture de gènes spécifiques qui se trouvent respectivement dans la région chromosomique 2q35 et 1p36, suivie d’une fusion. Les gènes impliqués dans les chromosomes 2 et 1 sont respectivement PAX3 et PAX7, qui codent pour des facteurs de transcription de la famille des boîtes appariées.3 Les gènes qui fusionnent avec PAX3 et PAX7 sont FOXO1 ou FKHR, qui sont situés sur le chromosome 13 et sont des membres de la famille des facteurs de transcription de la fourche. La fusion de ces gènes conduit à l’expression de protéines de fusion, qui agissent comme des activateurs transcriptionnels contribuant au développement des tumeurs en modifiant les voies de la croissance cellulaire et de l’apoptose, en modulant la différenciation myogénique et en stimulant la motilité et d’autres voies métastatiques.3,8 Au cours du développement normal, l’expression de PAX3 est nécessaire à la migration des précurseurs des cellules musculaires squelettiques vers les extrémités, tandis que l’expression de PAX7, un marqueur des cellules satellites des muscles squelettiques chez l’adulte, est nécessaire au renouvellement normal des cellules. Les deux protéines sont rapidement dégradées au cours de la différenciation myogénique précoce. Cependant, dans les SRA, la fusion de PAX3/PAX7 avec FOXO1 leur donne une demi-vie plus longue.7
La RT-PCR et l’hybridation in situ en fluorescence sont des méthodes qui ont été utilisées pour analyser la fréquence de la fusion de PAX3 et PAX7 dans les SRA (Figure 3). L’Intergroup Rhabdomyosarcoma Study (IRS), qui comprenait 78 cas de SRA, a constaté que 77 % des cas étaient positifs pour la fusion, 55 % des SRA exprimaient PAX3/FOXO1, 22 % exprimaient PAX7/FOXO1 et 23 % étaient négatifs pour la fusion9. Dans une étude publiée en 2010, nous avons trouvé une tendance similaire : dans 25 cas de SRA, nous avons constaté que 50 % étaient positifs pour PAX3/FOXO1, 3 % étaient positifs pour PAX7/FOXO1 et 30 % étaient négatifs pour le gène de fusion.10 Dans cette étude, 37 cas de SRA ont été analysés, dont 63 % étaient positifs pour PAX3/FOXO1, 5 % étaient positifs pour PAX7/FOXO1 et 32 % étaient négatifs pour la fusion, ce qui est cohérent avec d’autres rapports. La détection des protéines de fusion oncogènes, en particulier PAX3/FOXO1, a une valeur pronostique significative puisque le comportement tumoral biologique des RMS avec cette translocation est plus agressif, avec une mauvaise réponse au traitement et des récidives fréquentes. En revanche, les tumeurs présentant la translocation PAX7/FOXO1 ont une meilleure réponse au traitement, un stade clinique généralement plus bas et une survie plus longue11.
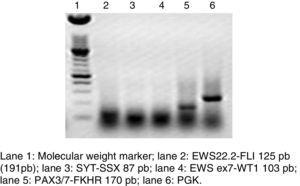
Électrophorèse sur gel d’agarose d’une RT-PCR, où est représentée la translocation PAX3/7-FKHR représentative du rhabdomyosarcome alvéolaire, écartant les autres néoplasmes solides.
Les mécanismes par lesquels la protéine chimérique PAX/FOXO1 contribue à l’oncogenèse des RMS ont été profondément étudiés. Apparemment, la quantité et la localisation cellulaire de la protéine sont critiques pour son activité oncogène. Les protéines PAX3/FOXO1 et PAX7/FOXO1 ont une activité transcriptionnelle 100 fois supérieure à celle des protéines sauvages PAX3 et PAX7.8,9 Les protéines PAX/FOXO1 sont exprimées par elles-mêmes à des niveaux élevés. La surexpression de PAX3 résulte d’une augmentation de la transcription indépendante du nombre de copies, tandis que la forte expression de PAX7 est associée à une amplification génique.12,13 Outre sa surexpression, PAX3/FOXO1 est significativement plus stable que PAX3, qui subit rapidement une protéolyse au cours de la différenciation musculaire.14 Au sein de la cellule, ces protéines chimériques peuvent se trouver dans le noyau ou dans le cytoplasme. Dans des conditions normales, la localisation du FOXO1 sauvage est contrôlée par AKT. Lorsque AKT est stimulé, il phosphoryle FOXO1, entraînant sa rétention dans le cytoplasme. Cependant, dans les ARMS, la protéine de fusion PAX/FOXO1 résiste à l’activité de l’AKT et reste principalement dans le noyau15. Une autre façon dont la protéine PAX/FOXO1 contribue à l’oncogenèse est en empêchant l’apoptose de la cellule tumorale par l’expression de gènes anti-apoptotiques comme Bcl-XL.
Le principal problème de la biologie des rhabdomyosarcomes est leur susceptibilité à se différencier indéfiniment en muscle squelettique, ce qui résulte d’altérations du programme myogénique, au niveau des kinases (c’est-à-dire p38 MAPK) et au niveau épigénétique. Au sein de ce dernier, on trouve le complexe polycomb-répressif ou JARID2, et les microARN (miRNA).16 Les miRNA sont une famille d’ARN non codifiants qui régulent l’expression des gènes au niveau post-transcriptionnel via l’inhibition ou la dégradation de l’ARN messager. Actuellement, on sait qu’un nombre important de miARN sont impliqués dans l’induction et la progression des rhabdomyosarcomes. L’expression de plusieurs miRNA est induite au cours du processus myogénique, et leurs cibles potentielles sont des gènes qui contrôlent la prolifération et la différenciation des myoblastes, entraînant une dérégulation de la prolifération cellulaire ou une différenciation myogénique aberrante. Les miARN les plus étudiés dans le muscle squelettique sont le miARN-1, le miARN-133a/b et le miARN-206, qui sont spécifiques au muscle, et le miARN-29b/c qui est exprimé de manière ubiquitaire.17
Il existe des preuves pointant vers la famille des miARN-29 comme suppresseurs de tumeurs puisque l’expression aberrante des membres de cette famille a été observée dans plusieurs types de cancer. La famille des miRNA-29s est une famille préservée, comprenant les miRNA-29a/b/c, qui joue un rôle important dans la prolifération cellulaire, l’apoptose, la migration et l’invasion.17-19 Le miRNA-29b est le membre de cette famille qui est exprimé à des niveaux plus élevés dans des conditions normales19.
Le pronostic des patients atteints de RMS dépend du grade de la tumeur, de l’âge, du type de résection, de l’histologie, de la présence des translocations mentionnées et du nombre de sites présentant des métastases.
La transition épithélio-mésenchyme (EMT) est un événement important pour l’invasion et les métastases de la tumeur, et elle est associée à un mauvais pronostic et à la chimiorésistance. L’EMT est un processus dans lequel les cellules épithéliales perdent leur polarité et l’adhésion cellule-cellule, et peuvent migrer et envahir d’autres organes20. Ce processus commence par la dissociation des jonctions intercellulaires (claudine, occludine, ZO-1, E-cadhérine et desmoplakine), ce qui entraîne la perte des microvillosités et de la polarité apicale-basolatérale.
Les cellules acquièrent une morphologie allongée, augmentent l’expression de l’α-actine des muscles lisses et renforcent leur capacité à migrer. Au dernier stade de l’EMT, les cellules acquièrent le potentiel de dégrader la membrane basale par l’expression de métalloprotéinases matricielles (MMP).20 L’EMT s’accompagne de changements moléculaires tels que la faible expression de la cytokératine et de la vimentine, la surexpression de la N-cadhérine et du facteur de transcription Snail (inhibiteur d’expression de la E-cadhérine).20
La chimiorésistance est l’un des principaux problèmes dans presque tous les types de cancer. Malgré le fait qu’il y ait une amélioration des agents chimiothérapeutiques, de nombreux patients atteints de cancer meurent parce qu’ils développent une chimiorésistance. Plusieurs études indiquent que les miARN sont impliqués dans la chimiorésistance,21 notamment le miARN-29b.
Le type de chimiothérapie que les patients atteints de RMS reçoivent dépend des facteurs de risque qu’ils présentent. Les patients à risque faible ou intermédiaire reçoivent de la vincristine, de la dactinomycine et du cyclophosphamide.22,23 Chez certains patients à risque intermédiaire, l’intensification de la dose de cyclophosphamide après la résection totale donne de bons résultats ; cependant, chez d’autres patients, l’intensification de la chimiothérapie n’améliore pas le résultat.24 Chez les patients à risque élevé, on utilise l’association ifosfamide-étoposide ou ifosfamide et doxorubicine. Ces patients ont généralement un mauvais pronostic. Il est intéressant de noter qu’il y a eu une bonne réponse au traitement chez les patients atteints de RMS à haut risque avec une ou plusieurs métastases. Ce type de SMR a un meilleur pronostic que les autres SMR métastatiques.25 Le SMRA à fusion négative se comporte de la même manière que le SMRE7. Nous avons constaté que l’expression du miRNA-29b est plus importante dans les tumeurs fusion-négatives, et qu’elles répondent mieux à la chimiothérapie.
Divulgation éthiqueProtection des sujets humains et animaux
Les auteurs déclarent que les procédures suivies étaient conformes aux règlements du comité d’éthique de la recherche clinique concerné et à ceux du Code d’éthique de l’Association médicale mondiale (Déclaration d’Helsinki).
Confidentialité des données
Les auteurs déclarent qu’aucune donnée de patient n’apparaît dans cet article.
Droit à la vie privée et au consentement éclairé.
0 commentaire