Introdução
p>Rabdomiossarcoma (RMS) é um tumor maligno de músculo estriado de origem mesenquimatosa considerado como o sarcoma de tecido mole mais recorrente em crianças e adolescentes, com uma incidência anual de 4,3 casos por milhão. Aproximadamente dois terços dos casos são diagnosticados em crianças (tipicamente por volta dos 6 anos de idade), e são ligeiramente mais comuns em homens, com uma relação macho/fêmea de 1,4:1.1 Devido à sua origem numa célula totipotente, os rabdomiossarcomas não ocorrem apenas no músculo esquelético, mas também noutros locais, tais como a cabeça, pescoço, tracto geniturinário e condutas biliares. Aproximadamente 40% dos RMS ocorrem na região da cabeça e pescoço, 20% na região geniturinária, 20% nas extremidades e os restantes 20% noutros locais.1
As variantes embrionárias e alveolares são os tipos histológicos mais frequentes, compreendendo 70 a 20% dos casos, respectivamente.2 O rabdomiossarcoma embrionário (ERMS) é o subtipo mais comum em lactentes e crianças pequenas e representa mais de dois terços de todo o RMS.1,3 Este tumor é composto por uma mistura suave de células fusiformes com áreas de estroma suave e solto, e está associado à perda de heterozigosidade no locus 11p15.3 Os tumores que estão localizados na cabeça, pescoço e região geniturinária apresentam frequentemente este tipo histológico.
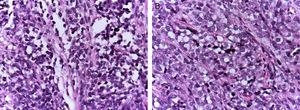
Rabdomyosarcomas alveolares (ARMS) aparecem geralmente na adolescência; estão tipicamente localizados nas extremidades e têm uma elevada capacidade de metástase.1,4 A sua histologia é caracterizada por um septo de tecido conjuntivo fibroso com células neoplásicas ligadas (Figura 1A), semelhante aos espaços alveolares observados no pulmão, onde algumas das células se destacam e ocupam o espaço. É composto de células uniformemente poligonais com núcleo redondo ou oval hipercrómico de grau elevado (Figura 1B).1 Ao contrário do ERMS, o ARMS apresenta dois tipos particulares de translocações cromossómicas: entre os cromossomas 2 e 13, t (2;13) (q35;q14), e entre os cromossomas 1 e 13, t (1;13) (p36;q14), que ocorrem em 80% dos casos.4 Estas alterações genéticas levam à fusão de duas famílias de factores de transcrição. A primeira está localizada nos cromossomas 1 ou 2 e envolve factores de transcrição PAX3 e PAX7, respectivamente. A família PAX de factores de transcrição é um grupo de genes envolvidos na diferenciação de órgãos e tecidos, e possui um domínio N-terminal de ligação de ADN, que inclui uma caixa emparelhada, motivos homeobox, e um domínio C-terminal de reactivação, enquanto que a segunda classe envolve membros da família de factores de transcrição da cabeça do garfo (FKHR) ou FOXO1.
>div>
(A) Aspecto histológico do rabdomiossarcoma alveolar: septos conjuntivos fibrosos estão a formar estruturas pseudo-alveolares, nas quais as células neoplásicas estão incrustadas. (B) Em algumas áreas, elas ocupam todo o espaço formando uma neoplasia sólida.
Os factores de transcrição das famílias PAX e FOXO1 possuem um domínio N-Terminal de união com o ADN e um domínio C-terminal de transativação. Os pontos de ruptura para PAX e FOXO1 ocorrem nos introns 7 e 1, respectivamente. Os genes fundidos codificam para duas proteínas quiméricas com actividade oncogénica, PAX3/FOXO1, e PAX7/FOXO1. Estas proteínas são compostas por um domínio 5′ de união ao ADN (PAX) e um domínio de transacção 3′ (FOXO1).4 Foi demonstrado que as transcrições PAX3/FOXO1 e PAX7/FOXO1 estão presentes em 55 e 22% do ARMS, respectivamente, enquanto que os restantes ARMS são negativos para o gene de fusão.5 Sabe-se que os ARMS com a translocação PAX7/FOXO1 têm um prognóstico muito mais favorável em comparação com aqueles que transportam a translocação PAX3/FOXO1. A mediana de sobrevivência de 4 anos para o primeiro é 75% e 8% para o segundo.6 Os doentes com ARMS que têm frequentemente metástases no diagnóstico têm uma sobrevivência mediana curta. Além disso, a presença de proteínas do gene de fusão PAX3/7-FOXO1 está associada a um prognóstico desfavorável.2
Caso clínico
Um paciente do sexo masculino de dois anos e três meses de idade, nativo de Yucatan, que não tinha antecedentes médicos significativos até cinco meses de idade, quando apareceu um volume violeta na sua narina esquerda. De acordo com os pais, foi tratado com quimioterapia e radioterapia numa clínica local, com remissão do tumor dois meses depois. Quando o rapaz tinha dois anos de idade, o tumor apareceu novamente, por isso vieram ao nosso hospital.
Ao exame físico, o seu peso era de 10,4kg e a sua altura de 100cm. Ele estava acordado, o seu olho direito tinha reflexo de luz pupilar normal, mas o olho esquerdo não era avaliável devido a um tumor localizado no meio do rosto, predominantemente do lado esquerdo, arroxeado, fetido, fixado a planos profundos. Os dentes foram deslocados. Não tinha linfadenopatias no pescoço. Não foram encontradas anomalias no tórax, abdómen ou membros.
A tomografia computorizada (TAC) mostrou um tumor lobulado, com margens bem definidas que envolveu desde a região frontal até ao chão maxilar. O diagnóstico histopatológico foi rabdomiossarcoma alveolar de fase IV com infiltração na medula óssea e líquido cefalorraquidiano. Foi tratado com antibióticos e começou a quimioterapia com adriamicina, actinomicina, ciclofosfamida, e vincristina. O primeiro ciclo foi concluído, e ele teve alta. Recebeu quatro ciclos de quimioterapia, com uma redução do tamanho do tumor de 50%. No último ciclo, cisplatina e irinotecan foram adicionados ao regime quimioterápico. O último TAC mostrou alargamento da cavidade nasal e antro maxilar com deformidade do lado esquerdo da face, deslocamento do globo ocular, infiltração na parede medial da órbita, turbinados mal definidos, paredes mediais e laterais do antro maxilar esquerdo e ocupação dos seios esfenoidais.
O quarto ciclo quimioterapêutico foi suspenso devido a febre e neutropenia, inchaço abdominal e diminuição do peristaltismo devido a hipocalemia e íleo metabólico. Um bacilo gram-negativo foi isolado em hemocultura. Uma sonda nasogástrica foi colocada, e a colite neutropénica foi descartada. A insuficiência respiratória levou a uma ventilação mecânica invasiva. O paciente teve desequilíbrio electrolítico e choque séptico refratário, e finalmente morreu.
Discussão
Rabdomiossarcoma é o sarcoma de tecido mole mais comum em crianças; é classificado em rabdomiossarcoma embrionário (ERMS), rabdomiossarcoma alveolar (ARMS), rabdomiossarcoma botrítico e rabdomiossarcoma de células fusiformes, com fenótipos e características clínicas diferentes (Figura 2). Foram recentemente descritas variantes menos comuns, como o rabdomiossarcoma esclerosante e as com características de rabdomiossarcoma. O ERMS e o ARMS são os mais prevalecentes e compreendem 70% e 20% dos casos, respectivamente. Destes, ARMS é o que tem pior prognóstico.2,3 Este comportamento tem sido associado à expressão de proteínas de fusão oncogénica resultantes de translocações cromossómicas, um mecanismo de tumorigenese comum a muitos tipos de cancro, incluindo um terço dos sarcomas.5
 br>>>/div>
br>>>/div>
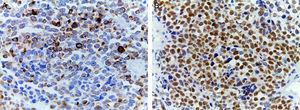
(A) Coloração imunohistoquímica mostrando positividade para desmin. Um filamento intermédio está presente no citoplasma das células musculares estriadas associadas às bandas Z. (B) Positividade nuclear intensa para a mioglobina, uma hemeproteína presente no músculo esquelético e que serve de portador e reservatório de oxigénio; aparece tardiamente na maturação sequencial do músculo e é positiva em 95% dos casos de rabdomiossarcomas.
A translocação cromossómica mais comum encontrada no ARMS é t(2;13) (q35;q14) PAX3/FOXO1, e a menos frequente é t(1;13) (p36;q14) PAX7/FOXO1.6,7 As translocações t(2;13) e t(1;13) resultam da decomposição de genes específicos que estão dentro da região cromossómica 2q35 e 1p36, respectivamente, seguidos de fusão. Os genes envolvidos nos cromossomas 2 e 1 são PAX3 e PAX7, respectivamente, que codificam os factores de transcrição da família da caixa emparelhada.3 Os genes que se fundem com PAX3 e PAX7 são FOXO1 ou FKHR, que estão localizados no cromossoma 13 e são membros da família da cabeça de forquilha dos factores de transcrição. A fusão destes genes leva à expressão de proteínas de fusão, que actuam como activadores transcripcionais que contribuem para o desenvolvimento tumoral alterando as vias de crescimento celular e apoptose, modulando a diferenciação miogénica, e estimulando a motilidade e outras vias metastáticas.3,8 Durante o desenvolvimento normal, a expressão PAX3 é necessária para a migração dos precursores das células musculares esqueléticas para as extremidades, enquanto que a expressão PAX7, um marcador de células satélites do músculo esquelético em adultos, é necessária para a rotação celular normal. Ambas as proteínas são rapidamente degradadas durante a diferenciação miogénica precoce. No entanto, no ARMS, a fusão de PAX3/PAX7 com FOXO1 dá-lhes uma semi-vida mais longa.7
RT-PCR e a hibridação fluorescente in situ são métodos que têm sido utilizados para analisar a frequência da fusão de PAX3 e PAX7 no ARMS (Figura 3). O Estudo Intergrupo Rhabdomyosarcoma (IRS), que incluiu 78 casos de ARMS, verificou que 77% dos casos eram positivos para a fusão, 55% dos ARMS expressaram PAX3/FOXO1, 22% expressaram PAX7/FOXO1, e 23% foram negativos para a fusão.9 Num estudo publicado em 2010, encontrámos uma tendência semelhante: em 25 casos ARMS, verificámos que 50% eram positivos para PAX3/FOXO1, 3% eram positivos para PAX7/FOXO1, e 30% eram negativos para o gene de fusão.10 Neste estudo, foram analisados 37 casos ARMS, 63% dos quais eram positivos para PAX3/FOXO1, 5% eram positivos para PAX7/FOXO1, e 32% eram negativos para a fusão, o que é consistente com outros relatórios. A detecção das proteínas de fusão oncogénica, particularmente PAX3/FOXO1, tem um valor prognóstico significativo, uma vez que o comportamento biológico do tumor em RMS com esta translocação é mais agressivo, com má resposta ao tratamento e recidivas frequentes. Em contraste, os tumores com a translocação PAX7/FOXO1 têm uma melhor resposta ao tratamento, uma fase clínica geralmente mais baixa, e uma sobrevivência mais longa.11
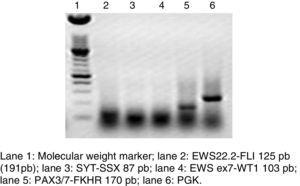
Agarose electroforese em gel de um RT-PCR, onde é mostrada a translocação representativa PAX3/7-FKHR do rabdomiossarcoma alveolar, descartando outras neoplasias sólidas.
Os mecanismos pelos quais a proteína quimérica PAX/FOXO1 contribui para a oncogénese do RMS foram profundamente estudados. Aparentemente, a quantidade e localização celular da proteína são fundamentais para a sua actividade oncogénica. Tanto PAX3/FOXO1 como PAX7/FOXO1 têm uma actividade transcripcional 100 vezes maior que as proteínas selvagens PAX3 e PAX7,8,9 As proteínas PAX/FOXO1 são expressas por elas próprias a níveis elevados. A sobreexpressão de PAX3 resulta de um aumento na transcrição que é independente do número de cópias, enquanto a alta expressão de PAX7 está associada à amplificação genética.12,13 Além da sua sobreexpressão, PAX3/FOXO1 é significativamente mais estável do que PAX3, que sofre rapidamente proteólise durante a diferenciação muscular.14 Dentro da célula, estas proteínas quiméricas podem ser encontradas no núcleo ou no citoplasma. Em condições normais, a localização do FOXO1 selvagem é controlada por AKT. Quando a AKT é estimulada, ela fosforila FOXO1, causando a sua retenção no citoplasma. Contudo, na ARMS, a proteína de fusão PAX/FOXO1 é resistente à actividade da AKT e permanece predominantemente no núcleo.15 Outra forma pela qual a proteína PAX/FOXO1 contribui na oncogénese é prevenindo a apoptose das células tumorais através da expressão de genes anti-apoptóticos como Bcl-XL.
O principal problema na biologia dos rabdomiossarcomas é a sua susceptibilidade de se diferenciarem em músculo esquelético indefinidamente, que é o resultado de alterações no programa miogénico, ao nível das kinases (i.e., p38 MAPK) e do nível epigenético. Dentro deste último, existem o complexo polycomb-repressivo ou JARID2, e os microRNAs (miRNAs).16 Os miRNAs são uma família de RNAs não codificadores que regulam a expressão genética a nível pós-transcritivo através da inibição ou degradação do RNA do mensageiro. Actualmente, existe um número significativo de miRNAs que se sabe estarem envolvidos na indução e progressão de rabdomiossarcomas. A expressão de vários miRNAs é induzida durante o processo miogénico, e os seus potenciais alvos são genes que controlam a proliferação e diferenciação dos miooblastos, resultando numa desregulamentação da proliferação celular ou numa diferenciação miogénica aberrante. Os miRNAs mais estudados no músculo esquelético são miRNA-1, miRNA-133a/b, e miRNA-206, que são específicos do músculo, e miRNA-29b/c que é expresso de forma omnipresente.17
Existem evidências que apontam para a família do miRNA-29 como supressores tumorais, uma vez que a expressão aberrante de membros desta família tem sido observada em vários tipos de cancro. A família miRNA-29s é uma família preservada, incluindo o miRNA-29a/b/c, que tem um papel importante na proliferação celular, apoptose, migração e invasão.17-19 miRNA-29b é o membro desta família que se expressa a níveis mais elevados em condições normais.19
O prognóstico dos pacientes com RMS depende do grau do tumor, idade, tipo de ressecção, histologia, presença das translocações mencionadas e número de sítios com metástases.
A transição epitelial-mesênquima (EMT) é um evento importante para a invasão e metástase do tumor, e está associada a um mau prognóstico e a quimiorresistência. O EMT é um processo em que as células epiteliais perdem a sua polaridade e a adesão celular, podendo migrar e invadir outros órgãos.20 Este processo começa com a dissociação das junções intercelulares (claudina, oclusina, ZO-1, E-cadherina, emoplakina), resultando na perda da polaridade microvelóide e apical-basolateral.
As células adquirem uma morfologia alongada, aumentam a expressão do músculo liso α-actin, e aumentam a sua capacidade de migrar. Na última fase do EMT, as células adquirem o potencial de degradar a membrana basal através da expressão de metaloproteinases matriciais (MMP).20 O EMT é acompanhado por alterações moleculares como a baixa expressão de citoqueratina e vimentina, a sobreexpressão de N-caderina e o factor de transcrição de caracol (inibidor da expressão de E-caderina).20
A quimioresistência é um dos principais problemas em quase todos os tipos de cancro. Apesar do facto de haver uma melhoria nos agentes quimioterápicos, muitos pacientes com cancro morrem porque desenvolvem quimiorresistência. Vários estudos indicam que os miRNAs estão envolvidos em quimioterapia,21 incluindo miRNA-29b.
O tipo de quimioterapia que os pacientes recebem com RMS depende dos factores de risco que apresentam. Os doentes de risco baixo ou intermédio recebem vincristina, dactinomicina e ciclofosfamida.22,23 Em alguns doentes de risco intermédio, a intensificação da dose de ciclofosfamida após ressecção total dá bons resultados; no entanto, noutros doentes, a intensificação da quimioterapia não melhora o resultado.24 Em doentes de alto risco, é utilizada a combinação de ifosfamida-etoposida ou ifosfamida-e doxorubicina. Estes pacientes têm geralmente um mau prognóstico. Curiosamente, tem havido uma boa resposta ao tratamento em pacientes com ERMS de alto risco com uma ou mais metástases. Este tipo de SGR tem um melhor prognóstico do que outros SGR metastáticos.25 O SGR fusion-negativo comporta-se de uma forma semelhante ao SGREM.7 Verificámos que a expressão de miRNA-29b é maior nos tumores fusion-negativos, e estes têm uma melhor resposta à quimioterapia.
Divulgação éticaProtecção de sujeitos humanos e animais
Os autores declaram que os procedimentos seguidos estavam de acordo com os regulamentos do comité de ética da investigação clínica relevante e com os do Código de Ética da Associação Médica Mundial (Declaração de Helsínquia).
Confidencialidade dos dados
Os autores declaram que não aparecem neste artigo quaisquer dados de doentes.
Direito à privacidade e consentimento informado
0 comentários