É bem conhecido que os pacientes com diabetes mellitus tipo 2 (T2DM) são caracterizados como tendo resistência à insulina, uma diminuição da absorção da glicose mediada pela insulina pelos tecidos periféricos (apesar dos níveis elevados de insulina) e taxas basais de gluconeogénese hepática excessivas.1,2 Uma deficiência na absorção periférica de glucose e a supressão da gluconeogénese contribuem ambas para a hiperglicemia pós-prandial (pós-prandial), enquanto que taxas basais excessivas de gluconeogénese hepática contribuem principalmente para o agravamento dos níveis de glicose de jejum. Até à data, a classe de medicamentos da classe biguanida, primarilysuprime as taxas basais excessivas de gluconeogénese que inclui primarilimetformina (Glucophage).3,4 A outra biguanida, a fenformina (Azucaps,Insoral, Fenformina), já não é aprovada pela FDA nos Estados Unidos devido a taxas inaceitáveis de acidose láctica, mas ainda pode ser utilizada e/ou adquirida por clínicos/pacientes noutros países.5
Porquê os diabéticos tipo 2 têm taxas excessivas na produção de glicose hepática basal?
Normalmente, a degradação do glicogénio e da gluconeogénese no fígado são ambos regulados em parte pela presença de insulina e têm um impacto directo no jejum dos níveis de glicose no sangue.1 No entanto, estando o T2DM num estado de resistência à insulina, a capacidade da insulina para activar as fosfatases proteicas, que desfosforilam a glicogénio fosforilase a e a glicogénio sintetase b que desliga a decomposição do glicogénio, é reduzida, permitindo assim que uma quantidade maior de glicogénio seja convertida em glicose 1-fosfato. Além disso, o estado de resistência à insulina pode também não ser suficiente para regular ou “abrandar” as duas etapas críticas ingluconeogénese que também coloca mais glicose no sangue. A primeira enzima sem regulação na resistência à insulina é a fosfenoenolpiruvaleroxicinase ((PEPCK); que converte o oxaloacetato em fosfenoolpiruvato) e a segunda é uma redução da quantidade de frutose 2,6-bisfosfato (F-2,6-P)produzida pela insulina que pode então inibir a enzima frutose 1,6-bisfosfatase. Todos os processos acima mencionados anormalmente regulados conduzem a uma maior quantidade de glucose 6-fosfato que pode então ser convertida de volta a toglucose no sangue através da glucose 6-fosfátase (uma enzima apenas encontrada no fígado).
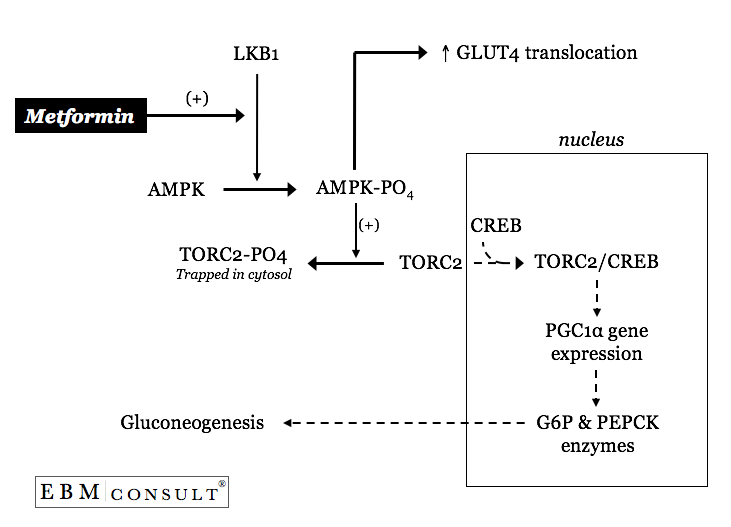
Como é que a metformina afecta um ou ambos estes processos anormalmente regulados na gluconeogénese hepática?
O benefício primário da metformina no T2DM tem sido na sua capacidade de “abrandar” as taxas basais aceleradas de gluconeogénese hepática sem efeito anaparente na rotação do lactato para a gluconeogénese ou aumentos na insulinsecreção.3,4 A metformina faz isto diminuindo a quantidade de carboxiquinase (PEPCK) e glucose 6-fosfatase (seefigure).6
p>Como é que faz isto?
Metformina pode activar uma cinase primária a montante chamada LKB1 resultando assim na fosforilação da proteína cinase activada por AMP (AMPK).7 A AMPK fosforilada resultará então no sequestro citosólico do factor de transcriçãoCREB denominado transdutor da CREBactividade 2 regulada (TORC2).7 Com a TORC2 agora presa no citosol do hepatócito (célula hepática) a CREB dentro do núcleo é agora nota eficiente na transcrição de um co-ativador transcritor chamado peroxisomeproliferador-activador receptor-g activado 1a (PGC1a).7 Com quantidades inferiores de PGC1a há menos activação transcripcional da glucose6-fosfatase e PEPCK, levando assim a um “abrandamento” das taxas basaisexcessivas de gluconeogénese hepática.7 Curiosamente, a activação de AMPK pela metformina também contribui para o controlo global da glucose, ao aumentar os aumentos mediados de AMPK na translocação dos transportadores de GLUT-4 inmuscle.8
Por isso, a metformina melhora os açúcares sanguíneos em jejum ao abrandar a gluconeogénese hepática basal “excessiva” sem alterações significativas nos níveis de insulina que se saberia causar hipoglicémia.3,4,9 As reduções médias nos níveis de glicemia em jejum e hemoglobina A1c enquanto onmetformina são aproximadamente 44-53 mg/dL (2,4-2,9 mmol/L)e 1,4-2%, respectivamente.3,4,9
- Monnier L, Colette C, Owens DR. Diabetes de tipo 2: uma diabetes bem caracterizada mas suboptimamente controlada. Podemos superar a divisão? Diabetes Metab. 2008;34(3):207-216.
li>Leiberman M, Marks AD, eds. Mark’s Basic Medical BiochemistryA Abordagem Clínica. 3ªEd. Philadelphia, PA: LippincottWilliams & Wilkins; 2009:479-566.li>Bristol-Myers Squibb Co. Glucophage (cloridrato de metformina) inserida na embalagem. Princeton, NJ; Agosto de 2008. Ligação obtida em 24.11.2008: Inserção da embalagem li>Cusi K, Consoli A, DeFronzo RA. Efeitos metabólicos da metformina sobre a glicose e o metabolismo do lactato de inoninina-dependente da diabetes mellitus. J Clin Endocrinol Metab 1996;81:4059-4067. Kumar A, Nugent K, Kalakunja A, Pirtle F. Acidose grave num doente com diabetes mellitus tipo 2diabetes, hipertensão e insuficiência renal. TÓRAX 2003;123:1726-1729. Mithieux G, Guignot L, Bordet J, Wiernsperger N. Mecanismos intra-hepáticos subjacentes ao efeito da metformina na diminuição da produção de glicose basal em ratos alimentados com um elevado teor de gordura. Diabetes 2002;51:139-143. li>Shaw RJ, Lamia KA, Vasquez D et al. A kinase LKB1 mede a homeostase da glucose no fígado e os efeitos terapêuticos da metformina. Science 2005;310(5754):1642-1646. li>Yamaguchi S. Katahira H, Ozawa S et al. Os activadores da proteína quinase activada por AMP aumentam a translocação do GLUT4 e a sua actividade glucosetransportes em adipócitos 3T3-LI. Am J Physiol Endocrinol Metab 2005;289(4):E643-E649. DeFronzo RA, Goodman AM,The Multicenter Metformin Study Group. Eficácia da metformina em doentes com diabetesmellitus não insulino-dependentes. N Engl J Med 1995;333:541-549. Nathan DM, Buse JB, Davidson MB et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from theAmerican Diabetes Association and the European Association for the Study ofDiabetes. Diabetes Care 2006;29(8):1963-1972.
0 comentários