Il est bien connu que les patients atteints de diabète sucré de type 2 (DT2) sont caractérisés par une résistance à l’insuline, une diminution de l’absorption du glucose médiée par l’insuline par les tissus périphériques (malgré des taux d’insuline élevés) et des taux basaux excessifs de gluconéogenèse hépatique.1,2 L’altération de l’absorption périphérique du glucose et la suppression de la gluconéogenèse contribuent toutes deux à aggraver l’hyperglycémie postprandiale (après les repas), tandis que les taux basaux excessifs de gluconéogenèse hépatique contribuent principalement à l’aggravation des taux de glucose à jeun. À ce jour, les médicaments de la classe des biguanides suppriment principalement les taux basaux excessifs de la gluconéogenèse, ce qui inclut principalement la metformine (Glucophage).3,4 L’autre biguanide, la phenformine (Azucaps, Insoral, Fenformin), n’est plus approuvé par la FDA aux États-Unis en raison de taux inacceptables d’acidose lactique, mais peut encore être utilisé et/ou acheté par les cliniciens/patients dans d’autres pays.5
Pourquoi les diabétiques de type 2 ont-ils des taux excessifs de production basale de glucose hépatique ?
Normalement, la dégradation du glycogène et la néoglucogenèse dans le foie sont toutes deuxen partie régulées par la présence d’insuline et ont un impact direct sur les taux de glucose sanguin à jeun1. Cependant, lorsque le DT2 est en état de résistance à l’insuline, la capacité de l’insuline à activer les protéines phosphatases, qui déphosphorylent la glycogène phosphorylase a et la glycogène synthase b qui bloquent la dégradation du glycogène, est réduite, permettant ainsi à une plus grande quantité de glycogène d’être convertie en glucose 1-phosphate. En outre, l’état de résistance à l’insuline peut également ne pas être efficace pour réguler ou « ralentir » les deux étapes critiques de la gluconéogenèse qui entraînent également une augmentation du glucose dans le sang. La première enzyme qui n’est pas régulée dans l’insulinorésistance est la phosphoénolpyruvatecarboxykinase (PEPCK), qui convertit l’oxaloacétate en phosphoénolpyruvate, et la seconde est une réduction de la quantité de fructose 2,6-bisphosphate (F-2,6-P) produite par l’insuline, qui peut alors inhiber l’enzyme fructose1,6-bisphosphatase. Tous les processus anormalement régulés ci-dessus conduisent à une plus grande quantité de glucose 6-phosphate qui peut ensuite être reconverti en glucose dans le sang via la glucose 6-phosphatase (une enzyme présente uniquement dans le foie).
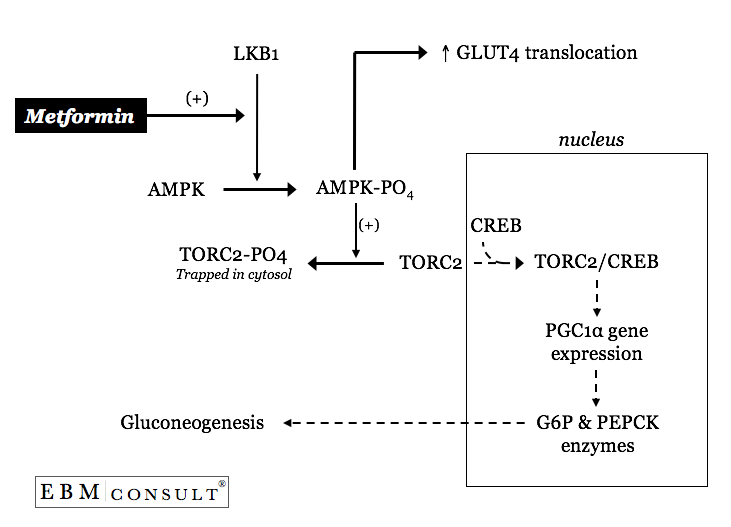
Comment alors la metformine affecte-t-elle l’un ou les deux de ces processus anormalement régulés dans la gluconéogenèse hépatique ?
Le principal avantage de la metformine dans le DT2 a résidé dans sa capacité à « ralentir » les taux basaux accélérés de la gluconéogenèse hépatique sans effet apparent sur le renouvellement du lactate pour la gluconéogenèse ou sur l’augmentation de la sécrétion d’insuline3,4. La metformine y parvient en diminuant la quantité d’enzymesphosphénolpyruvate carboxykinase (PEPCK) et glucose 6-phosphatase (voirfigure).6
Comment y parvient-elle ?
La metformine peut activer une kinase primaire en amont appelée LKB1 entraînant ainsi la phosphorylation de la protéine kinase activée par l’AMP (AMPK).7 L’AMPK phosphorylée entraînera alors le séquestre cytosolique du facteur de transcriptionCREB nommé transducteur de l’activité CREB régulée 2 (TORC2).TORC2 étant désormais piégé dans le cytosol de l’hépatocyte (cellule hépatique), CREB, dans le noyau, n’est plus efficace pour transcrire un cofacteur de transcription appelé coactivateur 1a du récepteur-g activé par le peroxysomeproliférateur (PGC1a).Avec des quantités plus faibles de PGC1a, il y a moins d’activation transcriptionnelle de la glucose6-phosphatase et de la PEPCK, ce qui conduit à un « ralentissement » des taux de base excessifs de la néoglucogenèse hépatique7. Il est intéressant de noter que l’activation de l’AMPK par la metformine contribue également au contrôle global du glucose en augmentant les augmentations médiées par l’AMPK de la translocation des transporteurs GLUT-4 dans le muscle.8
Par conséquent, la metformine améliore la glycémie à jeun en ralentissant la gluconéogenèse hépatique basale « excessive » sans modification significative des niveaux d’insuline qui seraient connus pour provoquer une hypoglycémie.3,4,9 Les réductions moyennes de la glycémie à jeun et de l’hémoglobine A1c sous metformine sont respectivement d’environ 44-53 mg/dL (2,4-2,9 mmol/L) et de 1,4-2 %.3,4,9
- Monnier L, Colette C, Owens DR. Le diabète de type 2 : une maladie bien caractérisée mais contrôlée de manière sous-optimale. Peut-on combler le fossé ? Diabetes Metab. 2008;34(3):207-216.
- Leiberman M, Marks AD, eds. Mark’s Basic Medical BiochemistryA Clinical Approach. 3rdEd. Philadelphie, PA : LippincottWilliams & Wilkins ; 2009:479-566.
- Bristol-Myers Squibb Co. Glucophage (chlorhydrate de metformine) notice d’emballage. Princeton, NJ ; août 2008. Lien obtenu le24/11/2008 : Package Insert
- Cusi K, Consoli A, DeFronzo RA. Effets métaboliques de la metformine sur le métabolisme du glucose et du lactate dans le diabète sucré insulino-dépendant. J Clin Endocrinol Metab 1996;81:4059-4067.
- Kumar A, Nugent K, Kalakunja A, Pirtle F. Acidose sévère chez un patient atteint de diabète sucré de type 2, d’hypertension et d’insuffisance rénale. THORAX 2003;123:1726-1729.
- Mithieux G, Guignot L, Bordet J, Wiernsperger N. Mécanismes intra-hépatiques sous-tendant l’effet de la metformine dans la diminution de la production basale de glucose chez les rats nourris avec un régime riche en graisses. Diabetes 2002;51:139-143.
- Shaw RJ, Lamia KA, Vasquez D et al. The kinase LKB1 mediatesglucose homeostasis in liver and therapeutics effects of metformin. Science 2005;310(5754):1642-1646.
- Yamaguchi S. Katahira H, Ozawa S et al. Les activateurs de la protéine kinase activée par l’AMP améliorent la translocation du GLUT4 et son activité de glucosetransport dans les adipocytes 3T3-LI. Am J Physiol Endocrinol Metab 2005;289(4):E643-E649.
- DeFronzo RA, Goodman AM,The Multicenter Metformin Study Group. Efficacité de la metformine chez les patients atteints de diabètemellitus non insulino-dépendant. N Engl J Med 1995;333:541-549.
- Nathan DM, Buse JB, Davidson MB et al. Management of hyperglycemia in type 2 diabetes : a consensus algorithmfor the initiation and adjustment of therapy : a consensus statement from theAmerican Diabetes Association and the European Association for the Study ofDiabetes. Diabetes Care 2006;29(8):1963-1972.
0 commentaire