Introduzione
Il rabdomiosarcoma (RMS) è un tumore maligno del muscolo striato di origine mesenchimale considerato il sarcoma dei tessuti molli più ricorrente nei bambini e negli adolescenti, con un’incidenza annuale di 4,3 casi per milione. Circa due terzi dei casi sono diagnosticati nei bambini (tipicamente intorno ai 6 anni di età), e sono leggermente più comuni nei maschi, con un rapporto maschio/femmina di 1,4:1.1 A causa della sua origine in una cellula totipotente, i rabdomiosarcomi non si verificano solo nel muscolo scheletrico ma in altre sedi, come la testa, il collo, il tratto genitourinario e i dotti biliari. Circa il 40% degli RMS si verifica nella regione della testa e del collo, il 20% nella regione genitourinaria, il 20% nelle estremità e il restante 20% in altre sedi.1
Le varianti embrionale e alveolare sono i tipi istologici più frequenti, comprendendo rispettivamente dal 70 al 20% dei casi.2 Il rabdomiosarcoma embrionale (ERMS) è il sottotipo più comune nei neonati e nei bambini piccoli e rappresenta più di due terzi di tutti i RMS.1,3 Questo tumore è composto da una miscela morbida di cellule fusiformi con aree di stroma morbido e sciolto, ed è associato alla perdita di eterozigosi al locus 11p15.3 I tumori localizzati nella testa, nel collo e nella regione genitourinaria presentano spesso questo tipo istologico.
I rabdomiosarcomi alveolari (ARMS) compaiono di solito nell’adolescenza; sono tipicamente localizzati nelle estremità e hanno un’elevata capacità di metastatizzare.1,4 La loro istologia è caratterizzata da un setto di tessuto connettivo fibroso con cellule neoplastiche attaccate (Figura 1A), simile agli spazi alveolari osservati nel polmone, dove alcune cellule si staccano e occupano lo spazio. È composto da cellule uniformemente poligonali con nucleo ipercromatico rotondo o ovale di alto grado (Figura 1B).1 A differenza delle ERMS, le ARMS presentano due particolari tipi di traslocazioni cromosomiche: tra i cromosomi 2 e 13, t (2;13) (q35;q14), e tra i cromosomi 1 e 13, t (1;13) (p36;q14), che si presentano nell’80% dei casi.4 Queste alterazioni genetiche portano alla fusione di due famiglie di fattori di trascrizione. La prima si trova sul cromosoma 1 o 2 e coinvolge i fattori di trascrizione PAX3 e PAX7, rispettivamente. La famiglia dei fattori di trascrizione PAX è un gruppo di geni coinvolti nella differenziazione di organi e tessuti, e possiede un dominio N-terminale di legame al DNA, che include una scatola accoppiata, motivi homeobox, e un dominio C-terminale di trans-attivazione, mentre la seconda classe coinvolge membri della famiglia dei fattori di trascrizione forkhead (FKHR) o FOXO1.
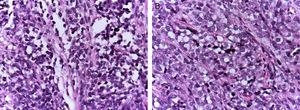
(A) Aspetto istologico del rabdomiosarcoma alveolare: i setti connettivi fibrosi formano strutture pseudo-alveolari, in cui le cellule neoplastiche sono incorporate. (B) In alcune aree, occupano l’intero spazio formando una neoplasia solida.
I fattori di trascrizione delle famiglie PAX e FOXO1 possiedono un dominio N-terminale di unione con il DNA e un dominio C-terminale di transattivazione. I punti di rottura per PAX e FOXO1 si verificano rispettivamente agli introni 7 e 1. I geni fusi codificano per due proteine chimeriche con attività oncogena, PAX3/FOXO1, e PAX7/FOXO1. Queste proteine sono composte da un dominio 5′ di unione al DNA (PAX) e un dominio 3′ di transattivazione (FOXO1).4 È stato dimostrato che i trascritti PAX3/FOXO1 e PAX7/FOXO1 sono presenti rispettivamente nel 55 e nel 22% delle ARMS, mentre le rimanenti ARMS sono negative per il gene di fusione.5 È noto che le ARMS con la traslocazione PAX7/FOXO1 hanno una prognosi molto più favorevole rispetto a quelle che portano la traslocazione PAX3/FOXO1. La sopravvivenza mediana a 4 anni per i primi è del 75% e dell’8% per i secondi.6 I pazienti con ARMS che spesso hanno metastasi alla diagnosi hanno una sopravvivenza mediana breve. Inoltre, la presenza di proteine del gene di fusione PAX3/7-FOXO1 è associata ad una prognosi sfavorevole.2
Caso clinico
Un paziente maschio di due anni e tre mesi di età, originario dello Yucatan, che non aveva una storia medica significativa fino a cinque mesi di età, quando una massa violetta apparve nella sua narice sinistra. Secondo i genitori, è stato trattato con chemioterapia e radioterapia in una clinica locale, con remissione del tumore due mesi dopo. Quando il bambino aveva due anni, il tumore è apparso di nuovo, così sono venuti al nostro ospedale.
All’esame fisico, il suo peso era di 10,4 kg e la sua altezza di 100 cm. Era sveglio, il suo occhio destro aveva un riflesso pupillare normale, ma l’occhio sinistro non era valutabile a causa di un tumore situato al centro del viso, prevalentemente sul lato sinistro, violaceo, fetido, fissato ai piani profondi. I denti erano spostati. Non aveva linfoadenopatie nel collo. Nessuna anomalia è stata trovata nel torace, nell’addome o negli arti.
La tomografia computerizzata (TC) ha mostrato un tumore lobato, con bordi ben definiti che ha coinvolto dalla regione frontale fino al pavimento mascellare. La diagnosi istopatologica era un rabdomiosarcoma alveolare al IV stadio con infiltrazione nel midollo osseo e nel liquido cerebrospinale. È stato gestito con antibiotici e ha iniziato la chemioterapia con adriamicina, actinomicina, ciclofosfamide e vincristina. Il primo ciclo è stato completato ed è stato dimesso. Ha ricevuto quattro cicli di chemioterapia, con una riduzione delle dimensioni del tumore del 50%. Nell’ultimo ciclo, al regime chemioterapico sono stati aggiunti cisplatino e irinotecan. L’ultima TAC ha mostrato un allargamento della cavità nasale e dell’antro mascellare con deformità del lato sinistro del viso, spostamento del bulbo oculare, infiltrazione della parete mediale dell’orbita, turbinati mal definiti, pareti mediali e laterali dell’antro mascellare sinistro e occupazione dei seni sfenoidali.
Il quarto ciclo chemioterapico fu sospeso per febbre e neutropenia, gonfiore addominale e diminuzione della peristalsi a causa di ipokaliemia e ileo metabolico. Un bacillo gram-negativo è stato isolato nell’emocoltura. Un tubo nasogastrico è stato collocato, e la colite neutropenica è stata scartata. L’insufficienza respiratoria ha portato alla ventilazione meccanica invasiva. Il paziente aveva uno squilibrio elettrolitico e uno shock settico refrattario, e alla fine è morto.
Discussione
Il rabdomiosarcoma è il più comune sarcoma dei tessuti molli nei bambini; è classificato in rabdomiosarcoma embrionale (ERMS), rabdomiosarcoma alveolare (ARMS), rabdomiosarcoma botrioide e rabdomiosarcoma a cellule fusate, con diversi fenotipi e caratteristiche cliniche (Figura 2). Varianti meno comuni sono state descritte recentemente, come il rabdomiosarcoma sclerosante e quelli con caratteristiche rabdoidi. L’ERMS e l’ARMS sono i più prevalenti e comprendono rispettivamente il 70% e il 20% dei casi. Di questi, l’ARMS è quello con la prognosi peggiore.2,3 Questo comportamento è stato associato all’espressione di proteine di fusione oncogeniche derivanti da traslocazioni cromosomiche, un meccanismo di tumorigenesi comune a molti tipi di cancro, compreso un terzo dei sarcomi.5
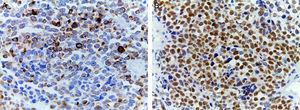 (A) Colorazione immunoistochimica che mostra positività per la desmina. Un filamento intermedio è presente nel citoplasma delle cellule muscolari striate associato alle bande Z. (B) Intensa positività nucleare per la mioglobina, un’emeproteina presente nel muscolo scheletrico e che serve come trasportatore e serbatoio di ossigeno; appare tardivamente nella maturazione muscolare sequenziale ed è positiva nel 95% dei casi di rabdomiosarcomi.
(A) Colorazione immunoistochimica che mostra positività per la desmina. Un filamento intermedio è presente nel citoplasma delle cellule muscolari striate associato alle bande Z. (B) Intensa positività nucleare per la mioglobina, un’emeproteina presente nel muscolo scheletrico e che serve come trasportatore e serbatoio di ossigeno; appare tardivamente nella maturazione muscolare sequenziale ed è positiva nel 95% dei casi di rabdomiosarcomi.
(A) Colorazione immunoistochimica che mostra la positività per la desmina. Un filamento intermedio è presente nel citoplasma delle cellule muscolari striate associate alle bande Z. (B) Intensa positività nucleare per la mioglobina, un’emeproteina presente nel muscolo scheletrico e che serve come trasportatore e serbatoio di ossigeno; appare tardivamente nella maturazione muscolare sequenziale ed è positiva nel 95% dei casi di rabdomiosarcomi.
La traslocazione cromosomica più comune trovata negli ARMS è t(2;13) (q35;q14) PAX3/FOXO1, e quella meno frequente è t(1;13) (p36;q14) PAX7/FOXO1.6,7 Le traslocazioni t(2;13) e t(1;13) risultano dalla rottura di geni specifici che si trovano rispettivamente nella regione cromosomica 2q35 e 1p36, seguita dalla fusione. I geni coinvolti nel cromosoma 2 e 1 sono rispettivamente PAX3 e PAX7, che codificano per fattori di trascrizione della famiglia paired box.3 I geni che si fondono con PAX3 e PAX7 sono FOXO1 o FKHR, che si trovano sul cromosoma 13 e sono membri della famiglia forkhead dei fattori di trascrizione. La fusione di questi geni porta all’espressione di proteine di fusione, che agiscono come attivatori trascrizionali che contribuiscono allo sviluppo del tumore alterando le vie della crescita cellulare e dell’apoptosi, modulando il differenziamento miogenico e stimolando la motilità e altre vie metastatiche.3,8 Durante lo sviluppo normale, l’espressione di PAX3 è richiesta per la migrazione dei precursori delle cellule muscolari scheletriche verso le estremità, mentre l’espressione di PAX7, un marker delle cellule satellite del muscolo scheletrico negli adulti, è richiesta per il normale ricambio cellulare. Entrambe le proteine sono rapidamente degradate durante la prima differenziazione miogenica. Tuttavia, nelle ARMS, la fusione di PAX3/PAX7 con FOXO1 dà loro un’emivita più lunga.7
RT-PCR e ibridazione in situ a fluorescenza sono metodi che sono stati utilizzati per analizzare la frequenza della fusione di PAX3 e PAX7 nelle ARMS (Figura 3). L’Intergroup Rhabdomyosarcoma Study (IRS), che includeva 78 casi di ARMS, ha scoperto che il 77% dei casi era positivo alla fusione, il 55% degli ARMS esprimeva PAX3/FOXO1, il 22% esprimeva PAX7/FOXO1 e il 23% era negativo alla fusione.9 In uno studio pubblicato nel 2010, abbiamo trovato una tendenza simile: in 25 casi di ARMS abbiamo trovato che il 50% era positivo per PAX3/FOXO1, il 3% era positivo per PAX7/FOXO1, e il 30% era negativo per il gene di fusione.10 In questo studio, sono stati analizzati 37 casi di ARMS, il 63% dei quali era positivo per PAX3/FOXO1, il 5% era positivo per PAX7/FOXO1, e il 32% era negativo per la fusione, che è coerente con altri rapporti. L’individuazione delle proteine di fusione oncogene, in particolare PAX3/FOXO1, ha un valore prognostico significativo poiché il comportamento biologico del tumore nel RMS con questa traslocazione è più aggressivo, con scarsa risposta al trattamento e frequenti recidive. Al contrario, i tumori con la traslocazione PAX7/FOXO1 hanno una migliore risposta al trattamento, uno stadio clinico solitamente più basso e una sopravvivenza più lunga.11
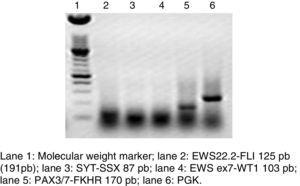
Elettroforesi su gel di agarosio di una RT-PCR, dove è mostrata la traslocazione rappresentativa PAX3/7-FKHR del rabdomiosarcoma alveolare, scartando altre neoplasie solide.
I meccanismi con cui la proteina chimerica PAX/FOXO1 contribuisce all’oncogenesi del RMS sono stati profondamente studiati. Apparentemente, la quantità e la localizzazione cellulare della proteina sono critiche per la sua attività oncogenica. Sia PAX3/FOXO1 che PAX7/FOXO1 hanno un’attività trascrizionale 100 volte maggiore delle proteine selvatiche PAX3 e PAX7.8,9 Le proteine PAX/FOXO1 sono espresse da sole ad alti livelli. La sovraespressione di PAX3 risulta da un aumento della trascrizione che è indipendente dal numero di copie, mentre l’alta espressione di PAX7 è associata all’amplificazione genica.12,13 A parte la sua sovraespressione, PAX3/FOXO1 è significativamente più stabile di PAX3, che subisce rapidamente la proteolisi durante il differenziamento muscolare.14 All’interno della cellula, queste proteine chimeriche possono essere trovate nel nucleo o nel citoplasma. In condizioni normali, la posizione del FOXO1 selvatico è controllata da AKT. Quando AKT è stimolata, fosforila FOXO1, causando la sua ritenzione nel citoplasma. Tuttavia, nella ARMS, la proteina di fusione PAX/FOXO1 è resistente all’attività di AKT e rimane prevalentemente nel nucleo.15 Un altro modo in cui la proteina PAX/FOXO1 contribuisce all’oncogenesi è impedendo l’apoptosi delle cellule tumorali attraverso l’espressione di geni anti-apoptotici come Bcl-XL.
Il problema principale nella biologia dei rabdomiosarcomi è la loro suscettibilità a differenziarsi indefinitamente in muscolo scheletrico, che è il risultato di alterazioni nel programma miogenico, a livello delle chinasi (per esempio, p38 MAPK) e dell’epigenetica. All’interno di quest’ultimo, ci sono il complesso polycomb-repressivo o JARID2, e i microRNA (miRNA).16 I miRNA sono una famiglia di RNA non codificanti che regolano l’espressione genica a livello post-trascrizionale attraverso l’inibizione o la degradazione dell’RNA messaggero. Attualmente, c’è un numero significativo di miRNA noti per essere coinvolti nell’induzione e nella progressione dei rabdomiosarcomi. L’espressione di diversi miRNA è indotta durante il processo miogenico, e i loro potenziali bersagli sono i geni che controllano la proliferazione e la differenziazione dei mioblasti, con conseguente deregolazione della proliferazione cellulare o una differenziazione miogenica aberrante. I miRNA più studiati nel muscolo scheletrico sono miRNA-1, miRNA-133a/b, e miRNA-206, che sono specifici per il muscolo, e miRNA-29b/c che è espresso ubiquitariamente.17
Ci sono prove che indicano la famiglia dei miRNA-29 come soppressori tumorali poiché l’espressione aberrante dei membri di questa famiglia è stata osservata in diversi tipi di cancro. La famiglia dei miRNA-29 è una famiglia conservata, che comprende i miRNA-29a/b/c, che ha un ruolo importante nella proliferazione cellulare, nell’apoptosi, nella migrazione e nell’invasione.17-19 Il miRNA-29b è il membro di questa famiglia che è espresso a livelli più alti in condizioni normali.19
La prognosi dei pazienti con RMS dipende dal grado del tumore, dall’età, dal tipo di resezione, dall’istologia, dalla presenza delle traslocazioni menzionate e dal numero di siti con metastasi.
La transizione epitelio-mesenchima (EMT) è un evento importante per l’invasione e le metastasi del tumore, ed è associata a una prognosi sfavorevole e alla chemioresistenza. L’EMT è un processo in cui le cellule epiteliali perdono la loro polarità e l’adesione cellula-cellula, e possono migrare e invadere altri organi.20 Questo processo inizia con la dissociazione delle giunzioni intercellulari (claudina, occludina, ZO-1, E-caderina e desmoplakin), con conseguente perdita dei microvilli e della polarità apicale-basolaterale.
Le cellule acquisiscono una morfologia allungata, aumentano l’espressione della muscolatura liscia α-actina e migliorano la loro capacità di migrare. Nell’ultima fase dell’EMT, le cellule acquisiscono il potenziale di degradare la membrana basale attraverso l’espressione delle metalloproteinasi di matrice (MMP).20 L’EMT è accompagnata da cambiamenti molecolari come la bassa espressione di citocheratina e vimentina, la sovraespressione di N-caderina e il fattore di trascrizione Snail (inibitore dell’espressione di E-caderina).20
La chemioresistenza è uno dei problemi principali in quasi tutti i tipi di cancro. Nonostante il fatto che ci sia un miglioramento degli agenti chemioterapici, molti pazienti con cancro muoiono perché sviluppano la chemioresistenza. Diversi studi indicano che i miRNA sono coinvolti nella chemioresistenza,21 compreso il miRNA-29b.
Il tipo di chemioterapia che i pazienti con RMS ricevono dipende dai fattori di rischio che presentano. I pazienti a basso o intermedio rischio ricevono vincristina, dactinomicina e ciclofosfamide.22,23 In alcuni pazienti a rischio intermedio, l’intensificazione della dose di ciclofosfamide dopo la resezione totale dà buoni risultati; tuttavia, in altri pazienti, l’intensificazione della chemioterapia non migliora il risultato.24 Nei pazienti ad alto rischio, viene utilizzata la combinazione di ifosfamide-etoposide o ifosfamide e doxorubicina. Questi pazienti hanno solitamente una prognosi sfavorevole. È interessante notare che c’è stata una buona risposta al trattamento nei pazienti con ERMS ad alto rischio con una o più metastasi. Questo tipo di SMR ha una prognosi migliore rispetto agli altri SMR metastatici.25 L’SMR negativo alla fusione si comporta in modo simile all’SMR.7 Abbiamo scoperto che l’espressione del miRNA-29b è maggiore nei tumori fusion-negativi, ed essi hanno una migliore risposta alla chemioterapia.
Discrezione eticaProtezione dei soggetti umani e animali
Gli autori dichiarano che le procedure seguite erano conformi alle norme del comitato etico di ricerca clinica pertinente e a quelle del codice etico dell’Associazione Medica Mondiale (Dichiarazione di Helsinki).
Confidenzialità dei dati
Gli autori dichiarano che nessun dato dei pazienti appare in questo articolo.
Diritto alla privacy e consenso informato
0 commenti