È noto che i pazienti con diabete mellito di tipo 2 (T2DM) sono caratterizzati da resistenza all’insulina, una diminuzione dell’assorbimento del glucosio mediato dall’insulina da parte dei tessuti periferici (nonostante gli elevati livelli di insulina) e tassi basali eccessivi di gluconeogenesi epatica.1,2 Una compromissione dell’assorbimento periferico del glucosio e la soppressione della gluconeogenesi contribuiscono entrambe al peggioramento dell’iperglicemia postprandiale (post-prandiale), mentre l’eccessivo tasso basale di gluconeogenesi epatica contribuisce principalmente al peggioramento dei livelli di glucosio a digiuno. Ad oggi, la classe dei farmaci biguanidi sopprime in primo luogo gli eccessivi tassi basali di gluconeogenesi, che comprende in primo luogo la metformina (Glucophage).3,4 L’altro biguanide, la fenformina (Azucaps, Insoral, Fenformin), non è più approvato dalla FDA negli Stati Uniti a causa dei tassi inaccettabili di acidosi lattica, ma può ancora essere usato e/o acquistato da medici/pazienti in altri paesi.5
Perché i diabetici di tipo 2 hanno tassi eccessivi nella produzione epatica basale di glucosio?
Normalmente, la degradazione del glicogeno e la gluconeogenesi nel fegato sono entrambe in parte regolate dalla presenza di insulina e hanno un impatto diretto sui livelli di glucosio nel sangue a colazione.1 Tuttavia, con T2DM essere in uno stato di resistenza all’insulina, la capacità di insulina per attivare le fosfatasi proteiche, che dephosphorylates glicogeno fosforilasi a e glicogeno sintasi b thatshut off ripartizione del glicogeno, è diminuita, permettendo così agreater quantità di glicogeno per essere convertito in glucosio 1-fosfato. Inoltre, lo stato di resistenza all’insulina può anche non essere asefficiente a regolare o “rallentare” le due fasi critiche ingluconeogenesi che mette anche più glucosio nel sangue. Il primoenzima mancanza di regolazione in insulino-resistenza è fosfoenolpiruvato-carbossilchinasi ((PEPCK); che converte ossalacetato in fosfoenolpiruvato) andthe secondo è una riduzione della quantità di fruttosio 2,6-bisfosfato (F-2,6-P) prodotto da insulina che può poi inibire l’enzima fruttosio1,6-bisfosfatasi. Tutti questi processi regolati in modo anomalo portano a una maggiore quantità di glucosio 6-fosfato che può essere riconvertito in glucosio nel sangue tramite la glucosio 6-fosfatasi (un enzima che si trova solo nel fegato).
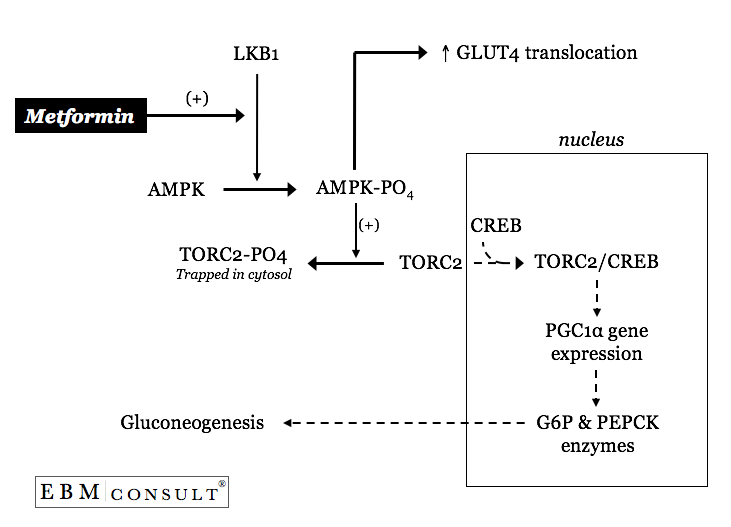
Come fa quindi la metformina a influenzare uno o entrambi questi processi regolati in modo anomalo nella gluconeogenesi epatica?
Il beneficio principale della metformina nel T2DM sta nella sua capacità di “rallentare” i tassi basali accelerati della gluconeogenesi epatica senza un effetto apparente sul turnover del lattato per la gluconeogenesi o sugli aumenti di insulinsecrezione.3,4 La metformina fa questo diminuendo la quantità di enzimi fosfoenolpiruvato carbossichinasi (PEPCK) e glucosio 6-fosfatasi (vedi figura).6
Come fa questo?
La metformina può attivare una chinasi primaria a monte chiamata LKB1, provocando così la fosforilazione dell’AMP-activated protein kinase (AMPK).7 L’AMPK fosforilata provocherà poi il sequestro citosolico del fattore di trascrizione CREB chiamato transducer of regulated CREBactivity 2 (TORC2).7 Con TORC2 ora intrappolato nel citosol dell’epatocita (cellula epatica) CREB all’interno del nucleo è ora non efficiente nel trascrivere un cofattore trascrizionale chiamato peroxisomeproliferator-activated receptor-g co-activator 1a (PGC1a).7 Con quantità inferiori di PGC1a c’è meno attivazione trascrizionale della glucosio6-fosfatasi e PEPCK, portando così a un “rallentamento” dei tassi basali eccessivi di gluconeogenesi epatica.7 È interessante notare che l’attivazione dell’AMPK da parte della metformina contribuisce anche al controllo generale del glucosio aumentando gli aumenti mediati dall’AMPK nella traslocazione dei trasportatori GLUT-4 nel muscolo.8
Quindi, la metformina migliora gli zuccheri nel sangue a digiuno rallentando la gluconeogenesi epatica basale “eccessiva” senza cambiamenti significativi nei livelli di insulina che sarebbero noti per causare ipoglicemia.3,4,9 Le riduzioni medie dei livelli di glucosio nel sangue a digiuno e dell’emoglobina A1c durante l’assunzione di metformina sono di circa 44-53 mg/dL (2,4-2,9 mmol/L) e 1,4-2%, rispettivamente.3,4,9
- Monnier L, Colette C, Owens DR. Diabete di tipo 2: una malattia ben caratterizzata ma non controllata in modo ottimale. Possiamo colmare il divario? Diabete Metab. 2008;34(3):207-216.
- Leiberman M, Marks AD, eds. Mark’s Basic Medical BiochemistryA approccio clinico. 3rdEd. Philadelphia, PA: LippincottWilliams & Wilkins; 2009:479-566.
- Bristol-Myers Squibb Co. Glucophage (metformina cloridrato) foglietto illustrativo. Princeton, NJ; agosto 2008. Link ottenuto il 24.11.2008: Inserto del pacchetto
- Cusi K, Consoli A, DeFronzo RA. Effetti metabolici della metformina sul metabolismo del glucosio e lattato innoninsulina-dipendente diabete mellito. J Clin Endocrinol Metab 1996;81:4059-4067.
- Kumar A, Nugent K, Kalakunja A, Pirtle F. grave acidosi in un paziente con tipo 2diabete mellito, ipertensione e insufficienza renale. PETTO 2003;123:1726-1729.
- Mithieux G, Guignot L, Bordet J, Wiernsperger N. Meccanismi intraepatici alla base dell’effetto della metformina nel diminuire la produzione basale di glucosio nei ratti alimentati con una dieta ricca di grassi. Diabete 2002;51:139-143.
- Shaw RJ, Lamia KA, Vasquez D et al. La chinasi LKB1 media l’omeostasi del glucosio nel fegato e gli effetti terapeutici della metformina. Science 2005;310(5754):1642-1646.
- Yamaguchi S. Katahira H, Ozawa S et al. Activators ofAMP-activated protein kinase enhance GLUT4 translocation and its glucosetransport activity in 3T3-LI adipocytes. Am J Physiol Endocrinol Metab 2005;289(4):E643-E649.
- DeFronzo RA, Goodman AM, The Multicenter Metformin Study Group. Efficacia della metformina in pazienti con diabete non insulino-dipendente. N Engl J Med 1995;333:541-549.
- Nathan DM, Buse JB, Davidson MB et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithmfor the initiation and adjustment of therapy: a consensus statement from theAmerican Diabetes Association and the European Association for the Study ofDiabetes. Diabetes Care 2006; 29(8):1963-1972.
0 commenti